| CGRP受容体拮抗薬 | CGRPを考えてみる | CSD | CSD -part2- |
| CSD -stroke2018- | ニトログリセリンと頭痛 | 若年者の慢性硬膜下血腫 | 飛行機頭痛 2012 |
| 薬物乱用頭痛の論文の紹介 | 慢性連日性頭痛 | 慢性疼痛の論文の紹介 | 睡眠時の頭痛 |
CGRP受容体拮抗薬2010/04/01-2012/12/01更新
片頭痛の治療はトリプタン製剤の出現により大きく変わりました。しかし、トリプタン製剤は、狭心症や心筋梗塞の既往のある患者様に禁忌とされています。また、トリプタン製剤が効果ない患者様もいます。このような場合にも、安心して使用できる薬剤の開発が望まれています。その一つが、CGRP受容体拮抗薬で、現在、欧米で開発中です。
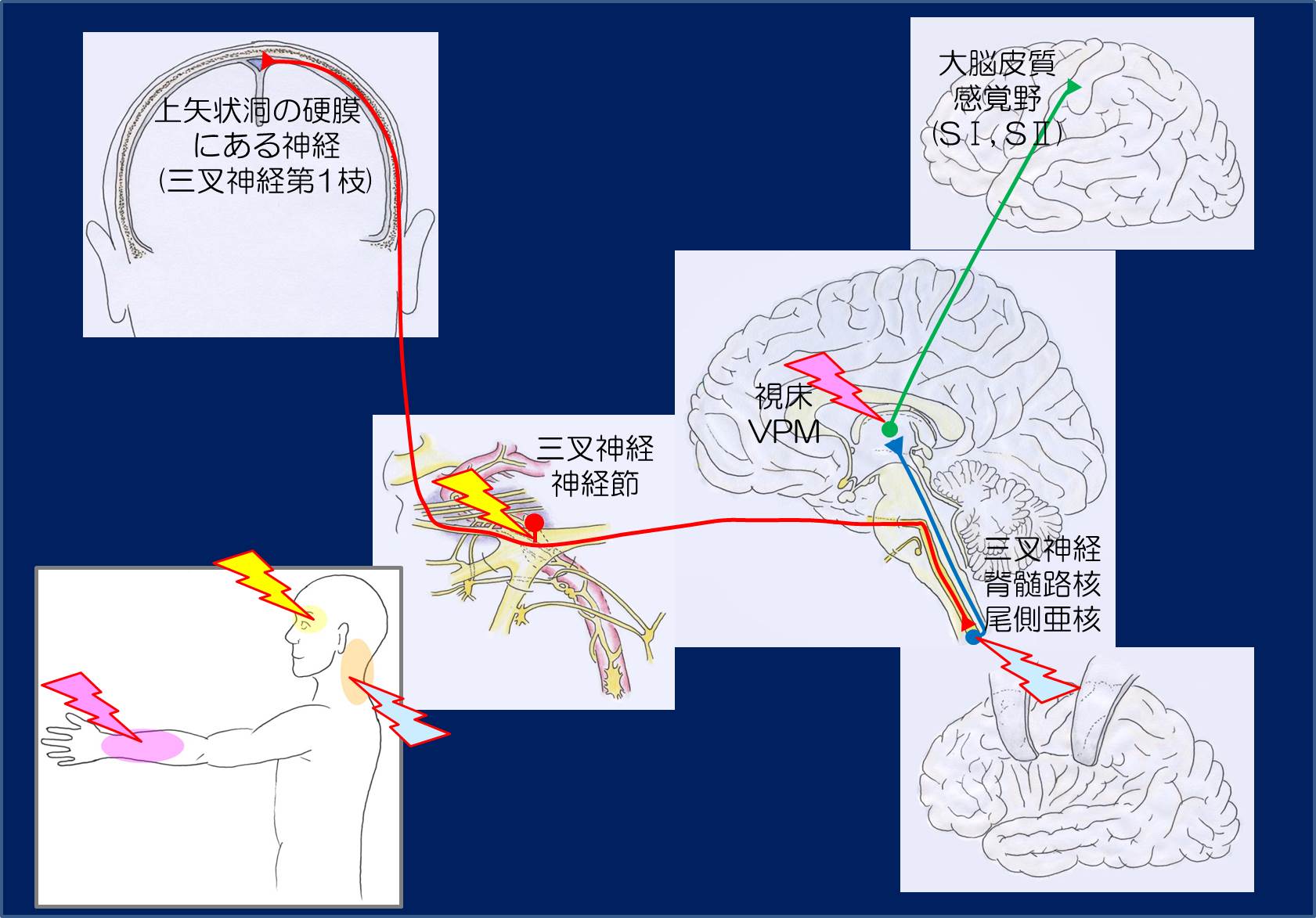
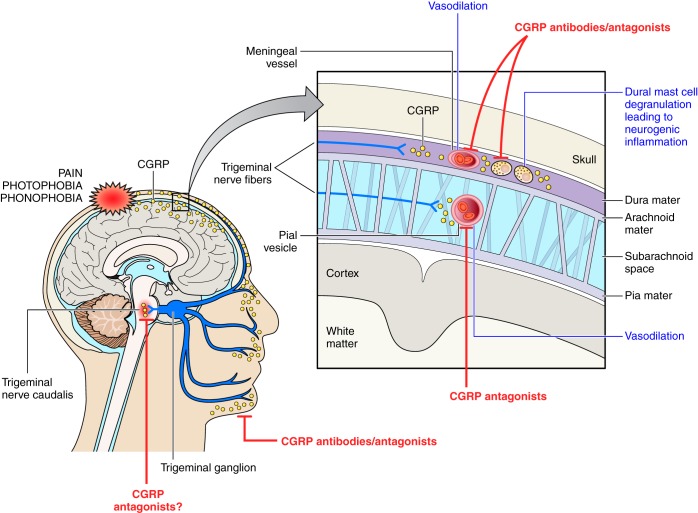
片頭痛の出現には、CGRPが重要な働きをしているといわれています。その働きには、次に示すように二つに大別されます。
- 三叉神経終末からCGRPが放出され、血漿タンパクが漏出し神経性炎症を惹起し、
血管拡張を引き起こすといわれています。(青で囲んだ部分) - 中枢神経系において、痛みの伝導に重要な役割を果たすと考えられています。
(赤で囲んだ部分)
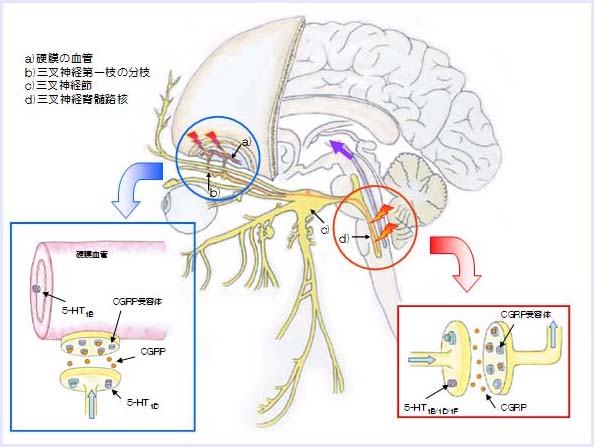
CGRP受容体拮抗薬は、これらの働きを阻害し、片頭痛を抑えると考えられ、特に中枢神経系に働くと考えられています。現時点では、虚血性心疾患を合併した片頭痛の患者様にも使用可能と考えられています。
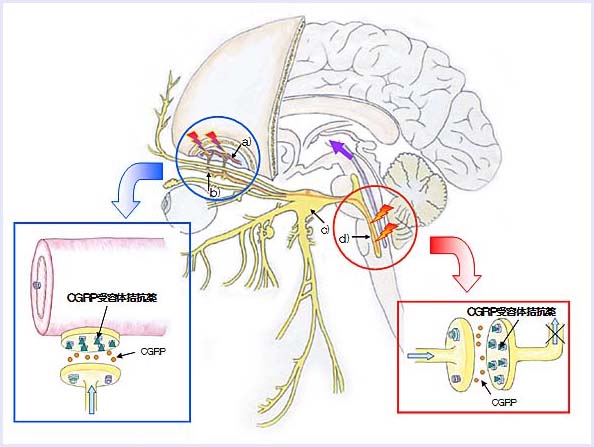
現在、世界で開発中の代表的なCGRP受容体拮抗薬には、オルケゲパントとテルカゲパントがあります。オルケゲパントは注射薬で、テルカゲパントは内服薬です。
テルカゲパント300mg製剤は、ゾルミトリプタン5mgと同様の効果があると考えられています。しかし、テルカゲパントは副作用として肝機能障害がみられ、その後の開発は中止されました。
開発中のCGRP受容体拮抗薬が他にもあるそうですが詳細なデータはまだ出ていないようです。
CGRPを考えてみる2017/05/20-2017/08/22更新
本HPでCGRPをとりあつかったのは2010年4月1日です。当時はCGRP受容体拮抗薬が使用できるようになるのではないかと期待されていました。
1988年に脳神経外科教室の神経化学グループで研究に携わっていた頃、CGRPという単語に遭遇したと記憶しています。その後の、知識としては、知覚に関係する、脳血管拡張に作用する、三叉神経が絡む、軸索反射、フレアなどでした。
くも膜下出血の脳動脈瘤の手術の際、内頚動脈に神経が絡み付いているのがみえるのですが(多分)、そのような薄い白い索状物を玉突き(手術器具)で、外してしまった方がよい(脳血管攣縮に対して)と指導を受けたことを思い出します(正しいのかどうかわかりませんし、そんなことはテキストに書きようもありません)。
現在、CGRP受容体拮抗剤の開発は上手くいっていないようですが、CGRP抗体やCGRP受容体抗体といった薬剤が新たに開発されています。現在の片頭痛治療のトピックスとなっており、ここで考えていきたいと思います。
2017年5月にCGRPを考えてみるを本HPにアップしました。その後、いくつかの論文を読みましたのでそのうちの二つを追加紹介します。
一つはEdvinsson先生のThe Trigeminovasucular Pathways: Role of CGRP and CGRP Receptors in Migraine という論文です。タイトルからして読みたくなる論文です。
もう一つは、Elucidating an Affective Pain Circuit That Creates a Threat Memoryという論文です。CGRPは、中枢神経系ではTrigeminovasucular Pathwaysにばかり注目集めているようですが、この論文では、PBN(parabrachial nucleus)から扁桃体(amygdala)に至る経路について概説しています。私は、今年4月から5月に酷い痛み発作に襲われたので興味深く読めました。
| 著者 | タイトル | |
| 1 | 小山なつ | 増補改訂新版 痛みと鎮痛の基礎知識 小山なつ著 2016年 技術評論社 p209 |
| 2 | 仙波恵美子 | CGRP(カルシトニン遺伝子関連ペプチド):クリニカルニューロサイエンス2012年2月号155-157 |
| 3 | Andrew F. Russo | CGRP: A New Target for Migraine.Annu Rev Pharmacol Toxicol.2015;55:533-552 |
| 4 | Smriti Iyengar | The role of CGRP in periphaeral and central mechanisms including migraine.Pain.2017;158:543-559 |
| 5 | F.A. Russel | CGRP:physiology and pathophysiology. Physiol Rev. 2014;94:1099-1142 |
| 6 | Edvinsson L. | The Trigeminovasucular Pathways: Role of CGRP and CGRP Receptors in Migraine.Headache.2017;57:47-55 |
| 7 | Hung S | Elucidating an Affective Pain Circuit That Creates a Threat Memory. Cell.2015;162:363-374 |
| CGRP序論① 小山なつ先生 | CGRP序論② 仙波恵美子先生 | CGRP review① Andrew F. Russo先生 | CGRP review② Smriti Iyengar先生 |
| CGRP review③ F.A. Russel先生 | CGRP2017 Edvinsson先生 | CGRPのもう一つの経路 Han先生 |
CGRP序論①
小山なつ先生2017/05/20
私は、小山なつ先生の【痛みと鎮痛の基礎知識】という本を理解できる範囲で読んできました。2016年12月に増補改訂されました。2冊の分冊が1冊となりました。
この中に、CGRPについての項があります。
わかりやすいので引用させていただきます。
増補改訂新版 痛みと鎮痛の基礎知識 小山なつ著 2016年 技術評論社 209ページより
CGRP(カルシトニン遺伝子関連ペプチド)は、中枢神経や一次求心性神経に存在する37個のアミノ酸からなるペプチドです。
カルシトニンとは骨からカルシウムの濃度調整を行う32個のアミノ酸からなるペプチドホルモンで甲状腺から分泌されます。カルシトニン遺伝子は組織特異的に選択的スプライシングを受け、同一のカルシトニン遺伝子から甲状腺ではカルシトニンが産生され、神経細胞ではCGRPが産生されます。
CGRPはSPを含有するDRGの小型ニューロンに共存していて、軸索反射により末梢終末からも放出されます。
血管拡張作用はSPよりも強く、フレアを生じさせ神経性炎症にも関与します。
さらに腕傍核から扁桃体へ投射するニューロンでも神経伝達物質としてCGRPが含まれることがわかってきました。
CGRPは炎症性疼痛時において、末梢だけでなく、扁桃体が関与する情動系にも関与し、痛みを過敏化させる可能性があります。
CGRPは、片頭痛だけではなく痛み全般にも関わっているようです。
CGRP序論②
仙波恵美子先生2017/05/20
私は、仙波恵美子先生の痛みに関する論文を理解できる範囲で読んできました。
CGRPについて、クリニカルニューロサイエンス2012年2月号155-157ページに記載されています。
わかりやすいので引用させていただきます。
CGRP(カルシトニン遺伝子関連ペプチド)は、37個のアミノ酸からなるペプチドで、3個のアミノ酸が異なるCGRPαとCGRPβの2つのアイソフォームが存在します。
小型-中型の後根神経節ニューロンはCGRPα、CGRPβの両方を発現し、大型のニューロンはCGRPαを発現するします。
CGRP陽性線維は皮膚の表皮や血管周囲に分布します。炎症性疼痛や神経障害性疼痛に関与すると考えられています。そして、CGRPが片頭痛にも関係があることを紹介しています。
CGRP review①
Andrew F. Russo先生2017/06/20
片頭痛に関するCGRPの論文は、膨大な量です。ひとつひとつ解決していくことは難しいので、手っ取り早くレビュー論文に飛びつくことにしました。
現在、地方の民間病院に勤務し、論文が簡単に入手できる状況ではないので、pubmedでfreeとなっている論文を参考にします。この中で探すのも大変です。
最初は、Andrew F. Russo先生のCGRP: A New Target for Migraine( Annu Rev Pharmacol Toxicol.2015;55:533-552)という論文です。
私がこの論文をはじめて読んだのは、2016年10月宮崎県の大崩山へ行く、博多-延岡の特急列車(特急と言ってもかなりゆっくりです)の中です。博多-延岡は列車で5時間くらいかかるので、ある意味、北海道よりも遠い?のです。晩秋の大崩山、最高でした。
イントロダクション:最初に片頭痛一般について述べてあるので省略します。
CGRPは、片頭痛の治療薬として魅力的なターゲットであると紹介されています。片頭痛は個人の損失ばかりでなく、広い意味で社会の損失でもあります。
それにもかかわらず、片頭痛の治療はトリプタン製剤(5-HT1B1D受容体アゴニスト)が1990年代に開発されて以降、大きな進歩はありません。敢えて挙げるとすれば、片頭痛予防薬の使用やボツリヌス毒素の使用などです。
現在のトリプタン製剤を中心とした片頭痛治療では、効果があるのは60%に過ぎず、また心血管系に対する副作用も考慮しなければなりません。
片頭痛と三叉神経血管系:片頭痛の原因は何か、この問題は未だに解決されていません。血管系に問題があるのか神経系に問題があるのか。
血管系のみで、片頭痛が生じることを説明することは難しいようです。脳幹への侵害受容信号にCGRPが関与していると考えられています。
CGRPとは
CGRPは、さまざまな機能をもつニューロペプチドです。
CGRPの免疫反応は、初期から中枢神経系・末梢神経系に存在することが知られており、心臓血管系や腸管系に関与すると考えられています。現在では、CGRPを含んだ神経は全身の主な臓器に至っています。片頭痛に対して、①心血管系、②神経原性炎症、③侵害受容に関与することが知られています。
CGRPは37個のアミノ酸からなり、6個のCGRP遺伝子ファミリーがあります。すなわち、カルシトニン、αCGRP、βCGRP、アミリン、アドレノメデュリン、アドレノメデュリン2(インターメディン)です。
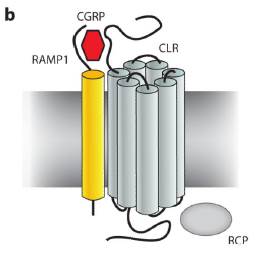
CGRP受容体を図に示します。CGRP受容体は、CLR、RAMP1、PCPの3つのコンポーネントから構成されます。CGRP受容体は、cAMPシグナル系を活性化させます。
CGRPは、CGRP関連ペプチドのアドレノメデュリン受容体、アミリン受容体にも結合します。アドレノメデュリン受容体は、CLRとRAMP2またはRAMP3から構成され、アミリン受容体はカルシトニン受容体とRAMP1から構成されます。これらの受容体は、三叉神経血管系に発見されています。特にCGRPによるアミリン受容体の活性化は、片頭痛に関係するのではないかと考えられています。
 |  |
| CGRPの37個のアミノ酸からなるCGRPの構造 | CGRP受容体構造で、CLR・RAMP1・PCPから構成されます。 |
 |
| 論文4では、CGRP受容体の構造と働きを示しています。 |
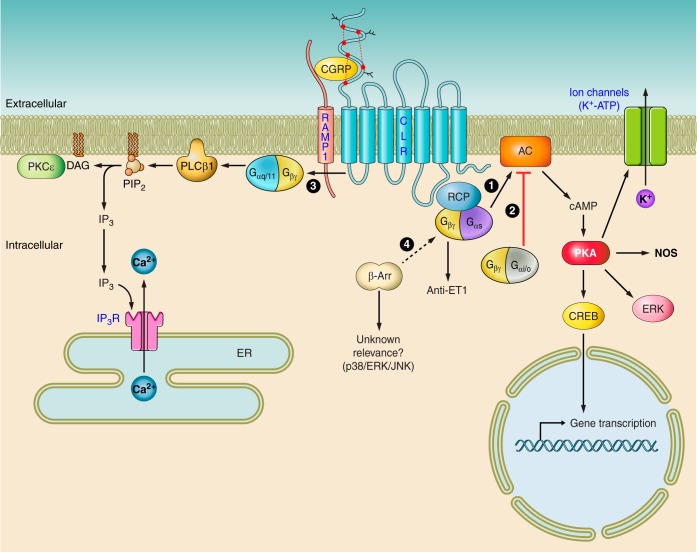 |
| 論文5からです。CGRP受容体の構造と働きを示しています。 |
片頭痛におけるCGRPのクリニカルエビデンス
流石にレビュー論文、新しい記載はありません。
この20年間、片頭痛の病態にCGRPが重要な役割を果たしていることが報告されてきました。
①片頭痛発作では、頚静脈でCGRPの血中濃度が上昇している。
そして、トリプタンの治療により、CGRP血中濃度は症状の消失とともに下降します。
しかし、CGRPの血中濃度は上昇しなかったという報告もあります。
②CGRPを静注すると片頭痛と同じよう症状が出現したという報告があります。
これは、片頭痛患者でのみみられ、片頭痛患者以外では即時性の頭痛を認めるのみです。
CGRPによる片頭痛様の症状は、NO、PACAPでもみられます。
③片頭痛に、CGRP受容体拮抗剤の効果が証明されています。
但し、CGRP受容体拮抗剤の作用部位は明らかではありません。何故なら、CGRP受容体拮抗剤は、中枢神経にまでは到達しないため、末梢で作用していると考えられています。但し中枢神経系で作用しているととの報告もあります。
トリプタンは、開発され20年が過ぎていますが、その作用機序は未だ明らかではありません。
片頭痛の治療のターゲットとして、CGRP
CGRP受容体拮抗剤
6個のCGRP受容体拮抗剤が開発されています。
1)オルケゲパント: 静注 66%で効果ありました(プラセボでは27%に効果あり)。しかし、経口タイプが開発されず尻すぼみとなったようです。
2) テルカゲパント: Merik社により開発された経口のCGRP受容体拮抗剤です。リザトリプタンやゾルミトリプタンなどと同様の鎮痛効果を有し、片頭痛の治療薬として期待されました。しかし、肝機能障害の副作用が認められ開発は中断されています。
次いでMK-3207というCGRP受容体拮抗剤が開発されましたが、これも同様に、効果はあるのですが副作用(肝機能障害)の点で開発は中断となっています。その他に3剤のCGRP受容体拮抗剤が開発された経緯がありますが、詳細な報告はされていないようです。
ここに至り、CGRP受容体拮抗剤の開発は進まなくなり、これに代わるかのようにCGRP抗体やCGRP受容体抗体の開発が活発となり、最近の頭痛学会でこれらの話題を耳にしないことはありません。
CGRP抗体とCGRP受容体抗体
これらの抗体の半減期は長く、片頭痛の予防薬としての可能性が高く、現在、4剤の開発が進んでいます。
1)CGRP抗体(ALD403, Alder Biopharmaceuticals):Phase1の結果から、安全性が確認され、発作性片頭痛の予防薬としての効果が確認されています。
2)CGRP抗体(LY2951742, Eli Lilly):Phase1及びPhase2の結果から、安全性および発作性片頭痛の予防薬としての効果が確認されています。
3)CGRP抗体(LBR-101, Labrys Biologics):Phase1で安全性が確認され、引き続きPhase1及び2で発作性片頭痛と慢性片頭痛に対する調査が行われています。この薬剤は慢性片頭痛もターゲットにしています。
4)CGRP受容体抗体(AMG334, Amgen):Phase1で安全性が確認され、引き続きPhase2の調査が計画されています。
このようにCGRP抗体やCGRP受容体抗体による片頭痛に対する治療(予防)は効果が認められています。CGRP抗体やCGRP受容体抗体は、脳血管関門(BBB)を通過しないと考えられおり、これらの薬剤の片頭痛に対する作用は、CGRPの末梢作用に対する効果と考えられています。別の考え方では、BBBのない領域へ関与しているかもしれません。
CGRPは片頭痛に対してどのように関与しているのでしょうか
CGRPの上昇は、片頭痛ではさまざまな感覚に対する過敏な反応に関与すると考えられます。CGRPが、末梢神経での侵害受容、中枢での感覚入力を修飾し、痛みに過敏になると考えられています。
末梢の求心性神経終末ではCGRPがリリースされ、各臓器の血管に働きかけます。
三叉神経のCGRPは、血管拡張、神経原性炎症、末梢性感作に関与します。
中枢では、CGRPおよびCGRP受容体は、光過敏(嫌悪)、中枢性感作、CSDに関与します。
脳血管拡張:末梢のCGRPの作用は、血管拡張です。CGRPは強力な血管拡張ペプチドとして知られ、脳血管や硬膜血管など拡張させます。CGRPによる血管拡張は、即時的です。しかし、CGRPによる片頭痛様の痛みは、遅発性であり、矛盾を感じます。
神経原性炎症と末梢性感作:CGRPは、神経原性炎症と末梢性感作に関与します。神経原性炎症、血管拡張、血漿蛋白漏出に関与します。血漿蛋白漏出に関しては、CGRPの直接作用というよりサブスタンスPやニューロキニンAなどによるを介した間接作用と考えられています。
CGRPの末梢での重要な作用に肥満細胞の脱顆粒があります。さまざまなサイトカインをリリースし、知覚神経を感作させます。これらには、CGRPによるプリン受容体P2X3やBDNFが関与していると考えられています。
視覚過敏(嫌悪):CGRPが光過敏(嫌悪)にも関与していることが報告されています。三叉神経血管系や視床、扁桃体など多くの部位での関与していると考えられています。
中枢性感作:CGRPは、グルタミン酸を介しニューロモデュレーターとしての役割を果たします。これは三叉神経の中枢性終末部や扁桃体で確認されています。
三叉神経脊髄路核で、CGRPは三叉神経中枢側からリリースされ、三叉神経脊髄路核から視床への二次ニューロンを修飾します。
ポストシナプスの脊髄視床ニューロンに存在するCGRP受容体は、グルタミン酸受容体(AMPA受容体やNMDA受容体)を介し機械的刺激に対するアロディニアに関与すると考えられています。
扁桃体と傍腕核の間のCGRP伝導は、中枢性感作や痛みに関する行動に関与すると考えられています。
CSD:CSDは、脳表からのKイオン、Hイオン、ATPなどにより、硬膜の侵害受容を刺激すると考えられています。CGRPは軟膜の神経終末よりリリースされ、神経原性炎症を惹起すると考えられています。
CGRP review②
Smriti Iyengar先生2017/07/12
2017年のレビュー論文です。内容は①とよく似ています。図がとてもクリアカットなので、図をみながら考えていきます。図を眺めているだけで筆者が何を考えているか、わかるような気がします。
受容体と機能に関する図1は、上記の受容体紹介の中段の図で紹介しました。
 |
| 図2 |
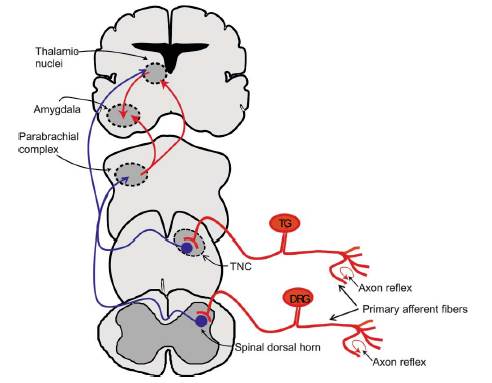
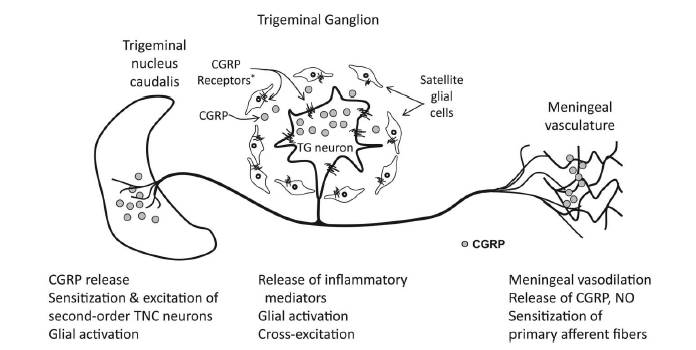
図2はCGRPの痛覚路に関連するものです。
CGRPを含んだものが赤い線で、含まないものが青い線で示されています。CGRPは、一次性求心路の中枢側の終末で発現しており、脊髄後角や三叉神経脊髄路尾状核で、二次ニューロンに信号を伝えています。
これはCGRPの感覚神経の働きとして知られていたのでよく理解できます。
もうひとつの赤い線で示される、傍腕核から扁桃体中心核(CeA)や視床への神経でもCGRPがみつかっています。これは小山先生が紹介していました。傍腕核や扁桃体が関与しているということは、痛みの情動系にも関与していることが考えられます。
細かいところでは、axon reflexにも関与しています。
図から離れますが、このレビュー論文では、中枢神経系におけるCGRPの分布やCGRP受容体の分布についても述べられており、紹介します。
CGRPは脊髄後角や三叉神経脊髄路核の高濃度に存在します。これは一次性求心性神経の中枢側の終末です。
このほかに、CGRPは心臓、冠動脈、血管床、腸管などの末梢神経に存在します。
一方、CGRP受容体は、脊髄後角に存在します。三叉神経では、小型-中型の神経に存在し、CGRP受容体は大型の神経、サテライトグリア細胞に存在します。
他の研究では、CGRP受容体は、硬膜血管に存在が証明されています。この部位は、三叉神経からの一次性求心性神経の末梢側の終末でもあり、CGRPの存在も確認されています。
最近の研究でもCGRPの受容体が三叉神経、硬膜血管の血管平滑筋に存在することが報告されています。
CGRPは、中枢神経系では、小脳、海馬、視床、大脳基底核、傍腕核、線条体、三叉神経脊髄路核、疑核などで存在が証明されています。
この中でも、傍腕核は重要であり、侵害受容信号を扁桃体(CeA)に伝え、ここでCGRPは発現しています。その他に傍腕核からは、rostal ventromedial medullaにも信号を送っています。
CGRPの受容体は、小脳・脳幹、硬膜に存在します。最近の研究では、松果体、乳頭体、median eminence、infundibular stem、PAG、背側縫線核、後視床下部に存在します。他の研究では、三叉神経脊髄路核、脊髄後角、薄束核、縫線核、最後野に存在します。
PAG、縫線核、薄束核などは、侵害受容に関係し、片頭痛に関係すると考えられている領域です。BBBのない領域も散見されますね。
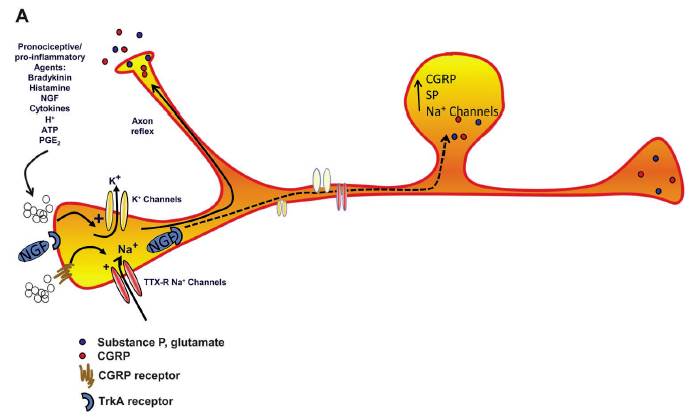 |
| 図3:末梢性感作 |
末梢性感作:末梢の組織や神経の損傷は、proinflammatory mediatorsをリリースします。
これらの物質は、ブラディキニン、ヒスタミン、NGF、サイトカイン、ATP、PGE2、H+などです。これらの物質は、末梢の侵害受容器やtrgger action potentialの閾値を下げます。この結果、侵害受容器を刺激します。
action potentialは、同じaxonの他の神経終末から、興奮性神経伝達物質をリリースします。これがaxonal reflexで、周囲の組織から炎症性物質をリリースし、周囲の神経終末を刺激します。このようにして炎症性反応が拡がっていきます。
持続した侵害受容器の刺激は、NGFのretrograde transport、受容体に結合し、TrkA-NGF complexによりtranscriptional changesを惹起し、NaチャンネルやCGRP、SPを増加させます。
これらの変化は、末梢神経の興奮性の増加、CGRP、SPのリリースの増加が末梢性感作を惹起・持続させます。
 |
| 図4:中枢性感作 |
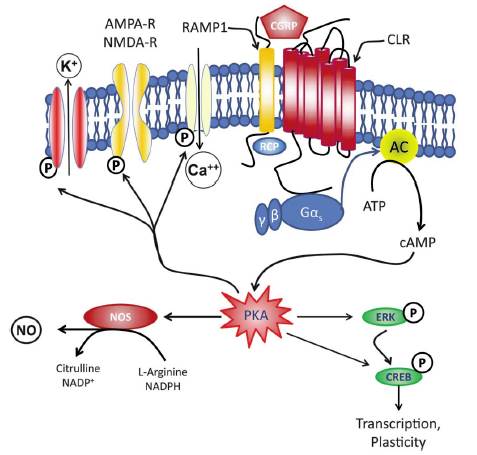
中枢性感作:一次性求心性入力が増加し、シナプス後ニューロンの興奮性が増加ます。NMDA受容体はカルシウム入力を増加させ、神経を感作に導きます。CGRP受容体はGs蛋白を活性化させ神経の興奮性を増加させます。
PKAおよびPKCのdownstream actvationは、NMDA受容体とCaイオンチャンネルのphosphorylationを進め神経興奮を増加させます。PKAは、NOS活性を増加、NOの増加を惹起します。
NO、Glutamate、PGE2は、retrograde transmitterとして振舞い、一次性求心性神経終末からの入力を増加させます。
さらにCGRPリリースの増加は、周囲のシナプス前のCGRP受容体を活性化しさらにtransmitterのリリースを増加させます。
PKAおよびPKCのphosphorylationの増加は、transcriptional change、受容体やイオンチャンネルのupregulationを惹起します。このようにして二次ニューロンの興奮性を高め中枢性感作を維持します。
とても興味深かったですね。感作について、以前、RCVS-真夏の夜の夢-のところで、Burstein先生の図とこれをmodifyし私が描いてみた図を紹介しましたが、ここで再び呈示します。比較してみてください。
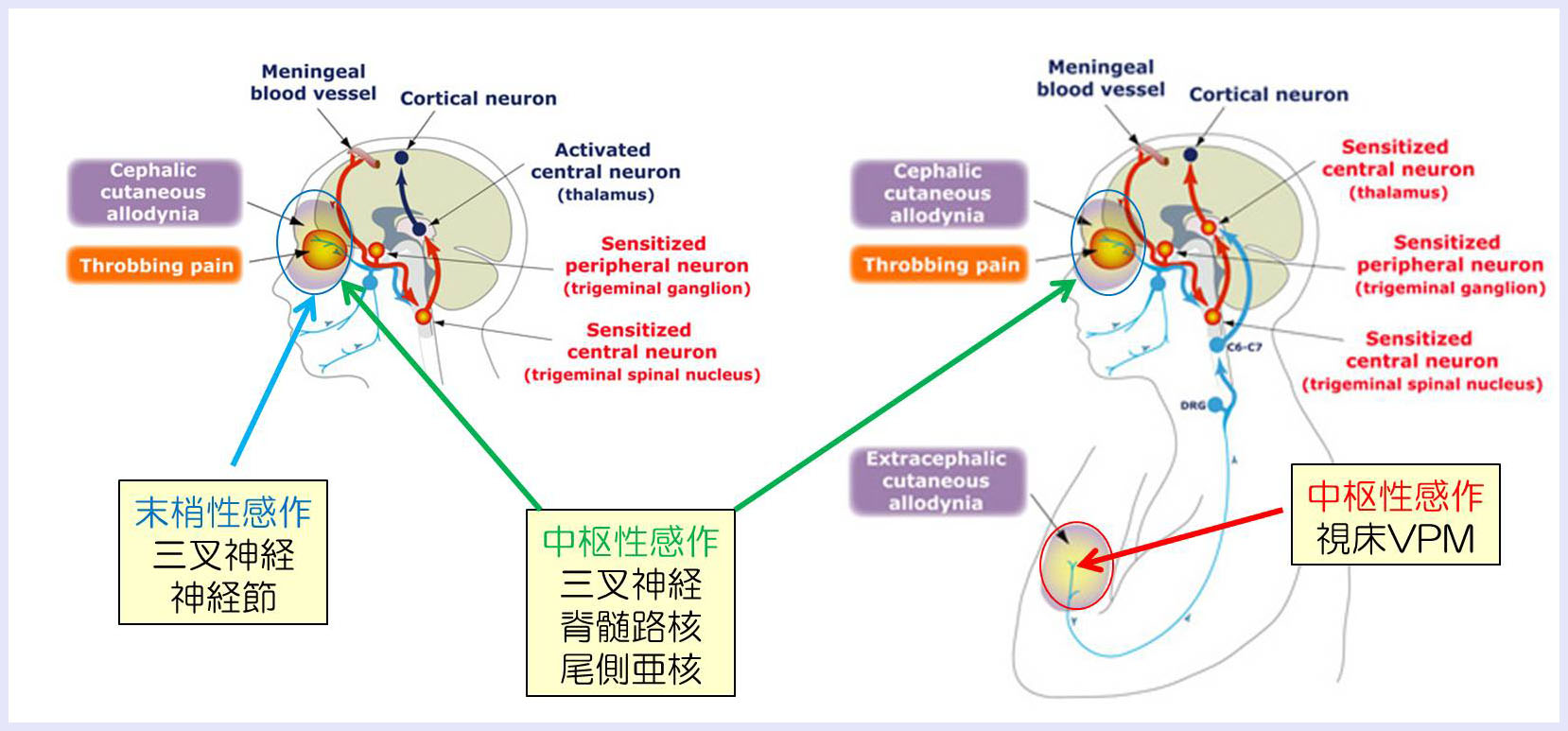 |
| Burstein先生らの提唱した概念です。 |
 |
| シェーマを作ってみました。三叉神経節が感作されると三叉神経第1枝領域に異和感、中枢(脊髄)まで感作されると後頚部まで、中枢(視床)まで感作されると異常感覚が上肢まで拡がります。 こんな感じだと思います。黄色、水色、桃色の色で対応しています。 |
 |
| 図5:説明なしでじっくり眺めましょう。 |
CGRP review③
F.A. Russel先生2017/07/12
CGRP:physiology and pathophysiology.というタイトルです。長文の大作です。出典は Physiol Rev. 2014;94:1099-1142からです。
長いので読めません。例によって図を眺めます。私がこの論文が良いと思ったのは、CGRPについては全身を考えなければいけないという視点です。図3からです。図1は構造・遺伝子、図2は受容体(上記で紹介)についてです。
 |
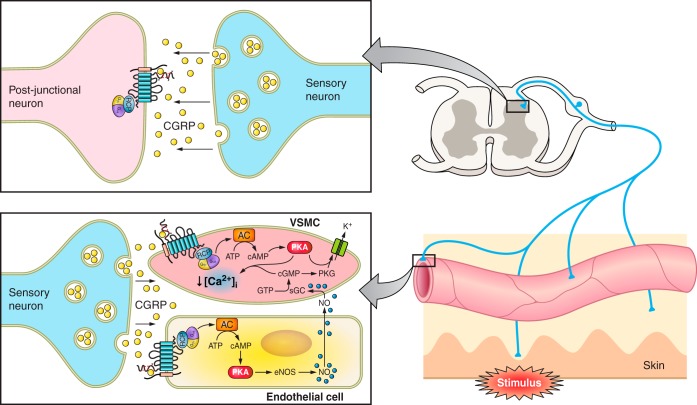
| 図3:CGRPの感覚神経と細動脈・皮膚への関与 |
図3では、CGRPの感覚神経と細動脈・皮膚への関与を示しています。前者は中枢性感作、後者は血管拡張でこれは血管平滑筋と血管内皮を介しています。
 |
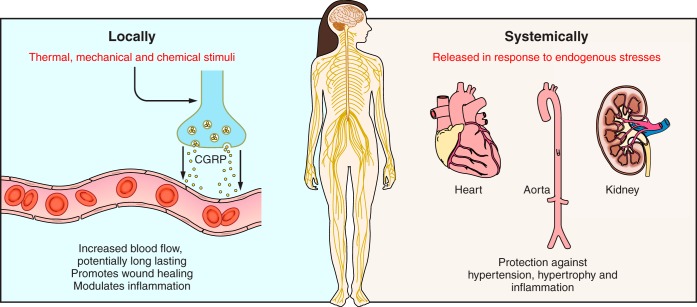
| 図4:CGRPの心血管系への関与(全身および局所の反応) |
図4では、CGRPの心血管系への関与を全身および局所の反応として示しています。
CGRPの局所の心血管系への反応として、血管拡張、血流量の増加、創治癒の改善が挙げられ、CGRPの全身の心血管系への反応として、高血圧の防御反応、炎症への防御反応などが挙げられています。
 |
| 図5:CGRPの片頭痛への関与 |
図5ではCGRPの片頭痛への関与が示されています。CGRPは中枢と末梢で関与していることが示されています。この論文(図)では、CGRP抗体は末梢で作用し、CGRP受容体拮抗剤は末梢ばかりでなく中枢にも作用しているのではないかと指摘しています(この内容は他の論文とは異なるようです)。
 |
| 図6:CGRPのいろいろな疾患への関与 |
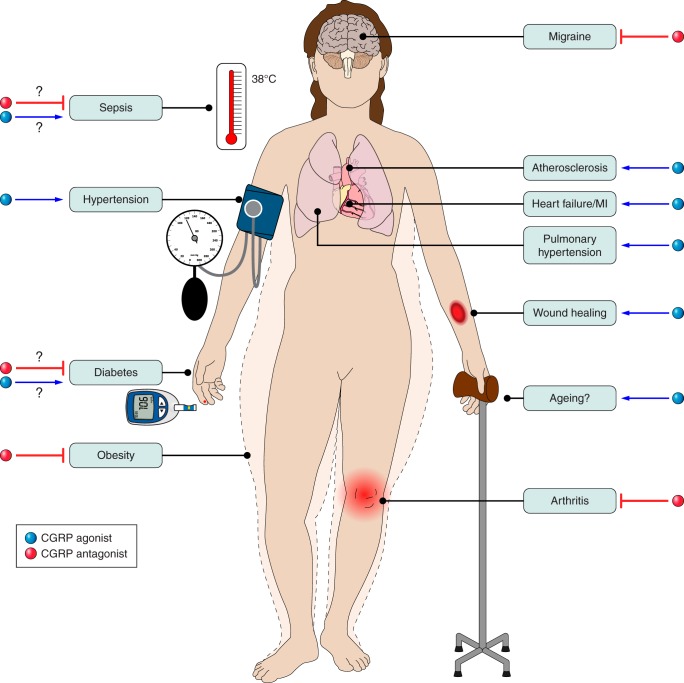
図6は、私にとってこの論文の最も大切なものです。CGRP作動薬(刺激薬)が治療薬となる疾患(高血圧、動脈硬化、心疾患、傷など)と、CGRP拮抗薬が治療薬となる疾患(片頭痛、関節炎、肥満)があることがわかります。ということは、各々が副作用になる危険性を秘めているのはないでしょうか。
ひとつの論文が優れていても、鵜呑みにしないことが大切だと思います。
CGRP2017
Edvinsson先生2017/08/22
Edvinsson先生が、The Trigeminovasucular Pathways: Role of CGRP and CGRP Receptors in Migraine.というタイトルで報告されています。
タイトルだけで、読まないわけにはいきません。内容はそれなりで、目新しいものはなかったようです。図が3つあります。本文と若干のずれもあるようですが、各々を紹介します。
 |
本HPでは、おなじみ(?)の図のようです。
 |
図のインパクトが小さいので、うすく色をつけてみました。
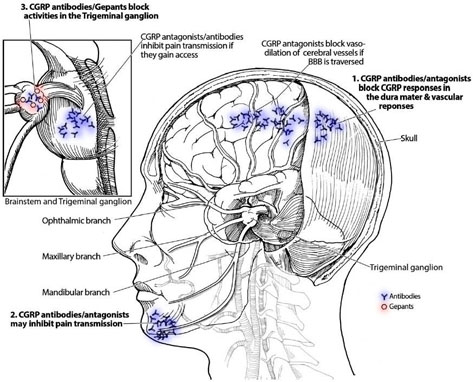
本文では、薬剤がBBBを通るか否かで、薬剤の作用部位を考察してあります。
CGRP受容体拮抗剤(gapant)やCGRP抗体やCGRP受容体抗体はいずれもBBBを通過しないので、作用部位が、BBBの外(硬膜血管や三叉神経節)、あるいはBBBの存在しない部位などが候補として挙げられています。
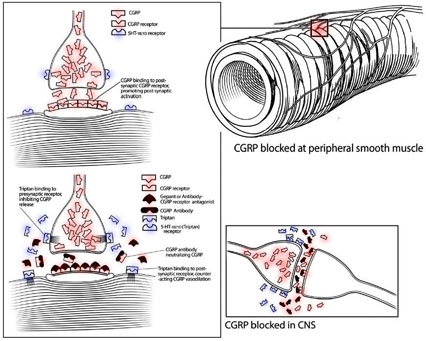 |
CGRP抗体やCGRP受容体抗体やトリプタンの作用部位が示されています。こんなものでしょう。
CGRPのもう一つの経路
Han先生2017/08/22
痛みは、①感覚と②嫌という二つにより成立します。①については上記の論文でCGRPが関与していることが明らかです。
しかし、小山先生の論文やSmriti Iyengar先生のCGRP review②論文で示されたように、CGRPは、PBから扁桃体への回路にも関与しているとことが示されています。
本論文では、この感覚(知覚)が嫌なもの、恐怖をきたすもの、として、CGRPを介したPBから扁桃体への経路が重要であることを実験により三段論法で証明した画期的な論文だと思います。
つまり、CGRPが知覚のみならず、情動にも関与していることを示した論文です。素晴らしい。
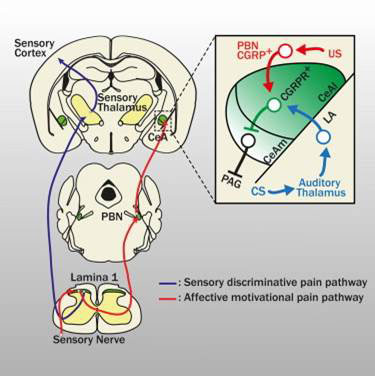 |
CSD2011/01/01
CSDは、cortical spreading depressionの略です。私は昭和60年に脳神経外科教室に入局しました。もう四分の一世紀も前のことです。当時の大学の脳神経外科教室には、生理グループ、神経化学グループ、脳虚血グループの三つの研究グループがあり、私は二番目の神経化学グループに入り研究しました。生理グループではCSDも研究していたようです。神経化学グループではエンドセリン(ET)とか心房性Naポリペプチド(ANP)などの受容体を研究し、私は脈絡叢におけるエンドセリン受容体について実験研究をしました。そういうわけで、私は、CSDには馴染みはなく、むしろ敬遠していました。
片頭痛の患者様を診察しますと前兆(視覚の異常)を訴える患者様が多くいます。CSDはこれらの前兆に深く関係があるといわれています。現在は、CSDの論文も読んでいます。いくつかの論文を読んでいると、”History of migraine with aura and cortical spreading depression from 1941 and onwards”(Cepharalgia 30:780-792, 2010)という論文がPC Tfelt-Hansenにより発表されました。これは、前兆のある片頭痛とCSDについて、1941年から現在までの論文を紐といて解説しています。この論文を柱に、必要があれば元の論文にもどって、自分の感想を追加しながらCSDと片頭痛について考えたいと思います。
必要文献を下の表に列記します。(8)がPC Tfelt-Hansenの文献で(1)から(7)は時代順に並べています。
Cortical spreading depression は皮質拡延性抑制と訳されています。日本語では何を表しているのかわかりません。皮質、これは脳表面のことで、拡延性は文字通り拡がっていくということです。何が拡がるかというと、【抑制】がです。つまり、【抑制が脳表面を拡がっていく】ということでしょうか。
| 著者 | タイトル | |
| 1 | Lashley KS | Patterns of cerebral integration induced by scotoma of migraine. Arch Neurol Psychiatry 1941;42:259-264 |
| 2 | Leao AAP | Spreading depression of activity in the cerebral cortex. J Neurophysiol 1944;7:359-360 |
| 3 | Olesen J | Focal heperemia followed by spreading oligemia and impaired activation of rCBF in classic maigraine. Ann Neurol 1981;9: 344-352 |
| 4 | Lauritzen M | Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol 1982; 12: 469-474 |
| 5 | Olesen J | Timing and topography of cerebral blood flow, aura, and headache during maigraine attacks. Ann Neurol 1990; 28: 791-798 |
| 6 | Lauritzen M | Cortical spreading depression in maigraine. Cepharalgia 2001; 21: 757-760 |
| 7 | Hadjikahani N | Mechanisms of migraine aura revealed by functional MRI in human visual cortex. PNAS 2001; 10:4687-4692 |
| 8 | PC Tfelt-Hansen | History of migraine with aura and cortical spreading depression from 1941 and onwards |
(1)1941年、Lashleyの論文から。
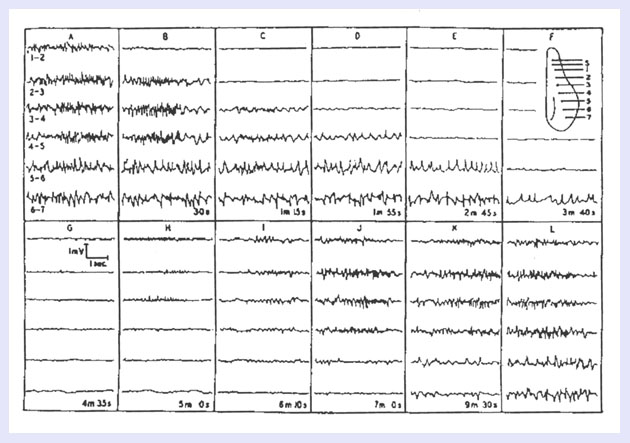
Lashleyは、視覚生理学者だそうです。彼が、自分の前兆のある片頭痛発作の閃輝暗点を鉛筆で克明に記録し、後頭葉(視覚野)にその原因があるのではないかと考えました。その図を示します。×印は固視点(固定点?)です。線状の印(彼の表現では要塞のようなという表現です)は閃輝を示し、点線の内部は暗点を示します。(閃輝は刺激状態を示し、暗点は抑制状態を示していると考えられています)。閃輝暗点が、中心から外側(側頭)の方向に拡がって行きます。それは両目の視野の一側に生じました。眼の問題であれば両目に出ることは少ないでしょうし、視野の一側に異常が出現することは、大脳の後頭葉の病変を示唆しています。
彼は、論文のまとめで、「前兆を伴う片頭痛で生じる閃輝暗点を短い間隔で記録し、後頭葉(視覚野)で1分間に3mmのスピードで興奮の波として拡がり、引き続き抑制の波が後頭葉(視覚野)を拡がっていき、同様に回復していく」と述べています。

この図からは、視野の異常は右なので、病変は左の後頭葉だと考えられます。
(2)CSDは、1944年にLeaoらによって発表されました。
Leaoらは、実験的けいれんの研究を行なっていました。脳表で、てんかん波(とがった波)を引き起こし、これが拡がっていくことを研究課題としました。この目的のために、麻酔をかけたウサギで、頭を開いて脳表を電気刺激し、脳表の6箇所の電極でその刺激の反応を拾うという実験です。てんかん波の拡がりを期待していたのですが、実際には、てんかん波は現れず、脳波は平坦となり、これは時間とともに脳表全体に拡がり、そしてその順番で回復していくという現象が現れました。この研究をもとに、その後、様々な分野でCSDが研究されています。
【(1)と(2)の論文の小括】Lashleyが1941年に自分の前兆のある片頭痛で、閃輝暗点を詳細に記載したこと(1)、Leaoらが、ウサギでCSDを示したこと(2)が、黎明期といえます。その後、1958年にMilnerが、この二つの事象を取り上げ、これら二つの事象が本当に関係しているのであれば、この事は片頭痛治療の改善に結びつくであろうと提唱しています。しかし、半世紀が過ぎた現在でもこのことは解明されていません。なにより、片頭痛の患者で実際にCSDが証明された報告はありません。
次に1967年頃より脳血流量測定が可能となり、片頭痛とCSDの研究が進みました。1981年のOlesenらの研究と1982年のLauritzenらの研究が有名です。Olesenは片頭痛の患者の脳血流量を測定し、Lauritzenは、ラットでCSDをつくり、脳血流量測定を行っています。当時、片頭痛の機序として、血管説(血管が縮み脳虚血を引き起こすために前兆が生じ、その後、血管が拡がり頭痛が生じるという考え方)が優勢でしたが、この事に一石を投じることになりました。
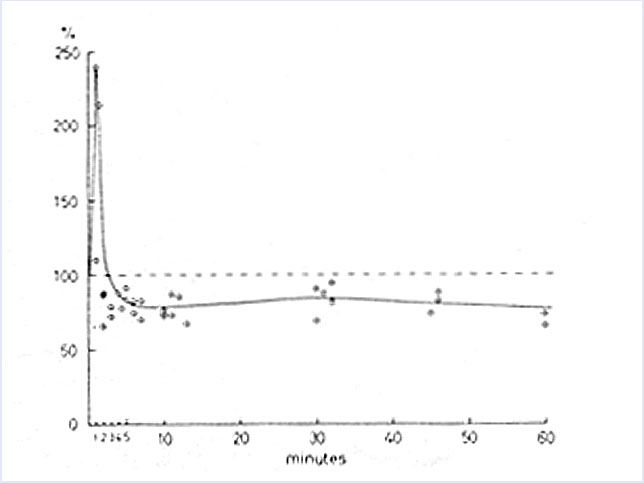
(3)1981年にOlesenは、6人の前兆を伴う片頭痛患者について、脳血流量を測定して報告をしています。
発作は局所の血流増加(hyperemia)から始まり、前兆期に脳血流量低下(oligemia)を認めています。この脳血流量低下は後頭葉から始まり15-45分で前方に拡がりました。そして頭痛は、既に脳血流量低下の時期に出現していることを明らかにしました。この事実から、Olesenらは大胆に片頭痛の血管説(血管が縮み脳虚血を引き起こすために前兆が生じ、その後、血管が拡がり頭痛が生じるという考え方)はあまりにも単純すぎると述べています。彼らは、神経機能の変化が前兆のある片頭痛発作に結びつくのではないかと考えました。
読後の感想:彼らは、1976-79年の4年間にいろいろな疾患の患者250例の脳血流量を測定しています(250例中14例が前兆のある片頭痛の患者です)。250例中、8例で脳血流量測定が低下しており、そのうち6例が、検査中に前兆のある頭痛を起こしたのです。片頭痛患者を集めて前向きに脳血流量測定をしたわけではないようです。脳血流量測定方法も現在の方法と異なり、頚動脈を直接穿刺し、そこから薬剤を注入して測定する方法です。現在では行なわれていない方法です。脳血管撮影で前兆のある片頭痛が誘発されるとは信じてもらえないのではないでしょうか、どうでしょう。1983年にLauritenらも同じ方法で、前兆のある片頭痛の脳血流測定を発表しており、そういうこともあるのかもしれません。
(4)1982年にLauritzenは、ラットで脳表にカリウムを滴下しCSDを生じさせ、その後、オートラジオグラフィー方法で経時的に脳血流量を測定しています。CSDが生じた後、瞬間的に脳血流量が増加し、その後に脳血流量が低下し持続しています。つまり、CSDが、脳血流量低下を起こすことを証明しました。
OlesenやLauritzenは、その後も多年にわたり片頭痛とCSDと脳血流量測定の研究を行なっています。
(5)Olesenの1990年の論文
Olesenのその後の研究のうち、1990年の論文を紹介します。(3)の論文の十年後です。この論文で、(3)の論文の考えが具現化されています。脳血流量測定の方法は、頚動脈穿刺による方法と現在行なわれているSPECTによる方法です。前者が計20例で、後者は43例です。後者は、前兆のある頭痛の誘発ができませんので、発作が起きたらできるだけ早くタクシーで通院してもらい検査するという方法です。
結果は図が全てを物語っています。この図は有名で、あたかも、片頭痛の脳血流量変化と誤解されていますが、この図はあくまで脳血管撮影で誘発された前兆のある片頭痛の患者の脳血流量変化のシェーマと考えるべきです。 大脳半球の後ろのほうで脳血流量が低下し、その後に前兆が出現します。頭痛は脳血流量低下の時期に出現します。脳血流量は増加していきますが、頭痛の性状はあまり変化しないということです。
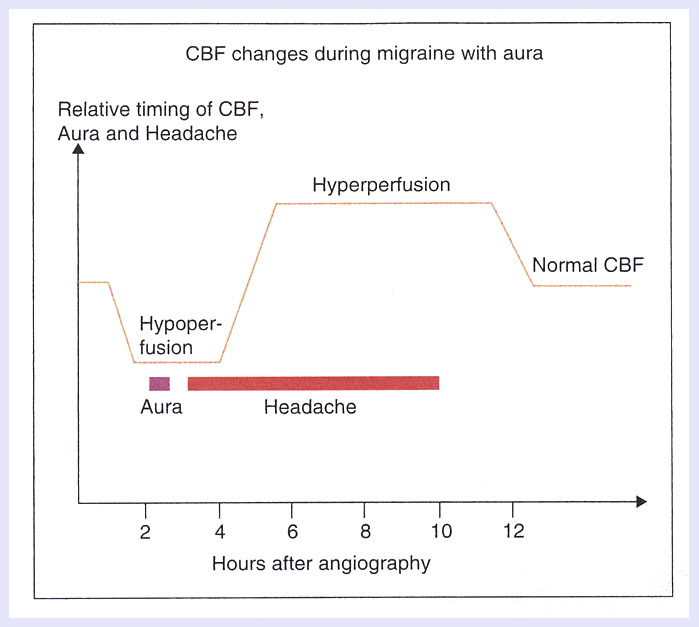
(6)Lauritzenは、1982年の発表以降、いろいろな論文を発表し、20年後の2001年に集大成というべき論文を発表しています。その中で有名なCSDによる片頭痛の発生仮説の図も、記載されているので、簡単に紹介します。この3個の模式図は、CSDの発作後の経時的変化をあらわしています。大まかには30分間隔でしょうか。
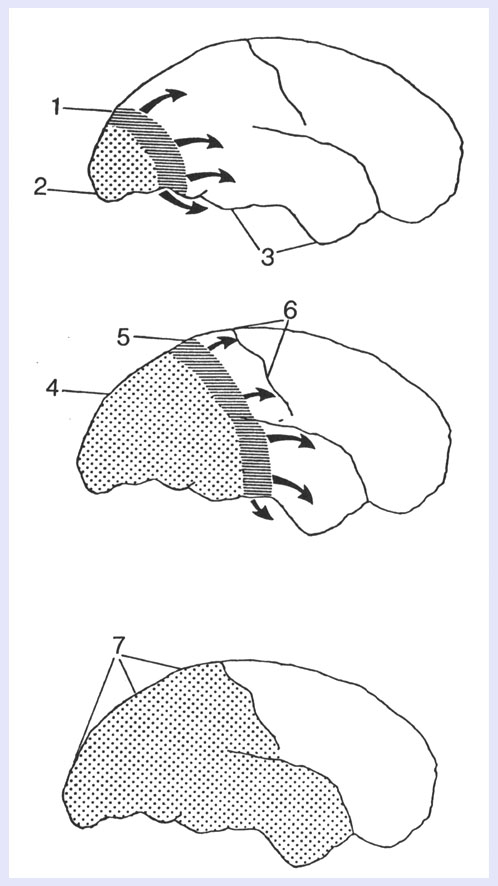
- 後頭葉でCSDが生じると、これは、脳の外側、内側、腹側を前方へ拡がって行きます。
- CSDに続き、後頭葉での脳血流減少が生じます。この減少の程度は20-30%で2-6時間継続します。
この程度の脳血流減少では脳虚血は生じません。 - CSDが関わっていない領域では、脳血流に変化は見られません。
- CSDが前方に拡がっていき、それに伴い、脳血流の低下した領域は拡大していきます。
- CSDが中心後野の感覚野に達すると、感覚に関わる症状が出現します。
- CSDは、通常は中心溝に到達すると停止します。多くの場合、これ以上は拡がりません。
腹側に拡がったCSDは痛覚-感受性線維を活性化させ頭痛を惹起します - CSDは停止し、脳血流の低下が継続します。この時期には、眼の症状や感覚の症状はなく、頭痛が主症状となります。
OlesenやLaurizenらの報告のあとは、MRIやPETによる検査でのCSDの解明が行なわれています。
現在では、MRI検査により、片頭痛の前兆と大脳皮質との活動性を報告がされています。
(7)Hadjikhniらは、fMRIを用いて前兆を伴った片頭痛の機序について報告しています。
Blood oxygenation level-dependent(BOLD)の信号を用いています。3人の患者で計5回の発作を調べています。一人は、バスケットボールなどの運動で発作が起こることがわかっていたので、片頭痛に伴う前兆の前、前兆の時期、そして頭痛の時期について経時的に調べることが可能でした。片頭痛のない時は、BOLD信号は左右対称で、正常の反応を示しています。左視野に起きた閃輝暗点をしめす前兆の発作時には、右後頭葉でBOLD信号は一過性に上昇し、その後に減少します。上昇はscintillationsの時期に一致すると考えられ、減少は暗点の時期に一致するものと考えられます。その後、徐々に回復していきます(1-B、2-B)。その他の二名は、MRI施設内で働いていたので、比較的はやく検査ができたようです。しかし、前兆の起こる前からの検査はできず、前兆が起きてからの検査となります(2-C)。
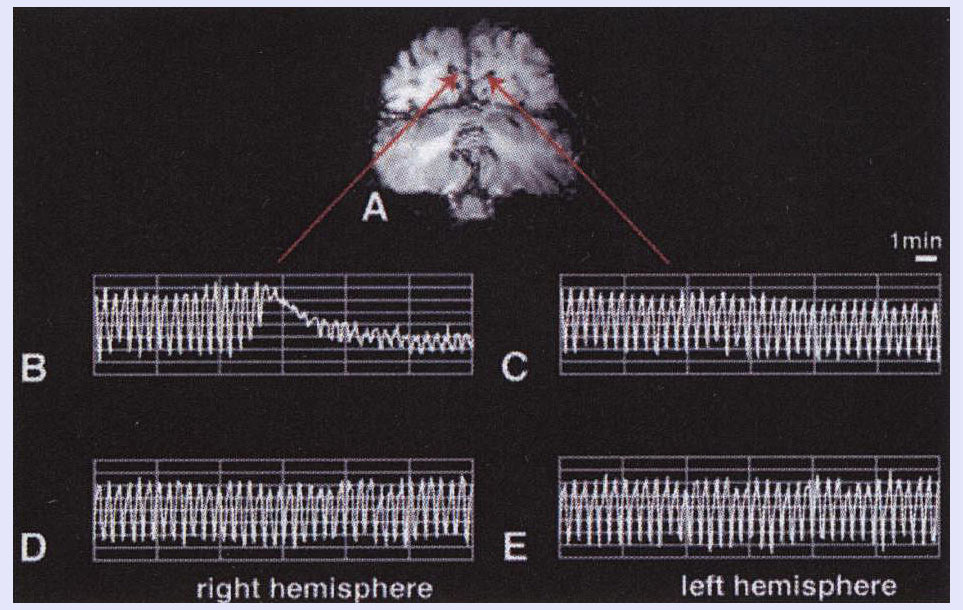
図1(バスケットボールで片頭痛発作を起こす患者のデータで、片頭痛発作前から検査が可能。B:左視野に起きた閃輝暗点を示す前兆の発作時に、右後頭葉でBOLD信号は一過性に上昇し、その後に減少しています。C:発作時には左後頭葉では変化なし。D&E:非発作時には変化なし。)
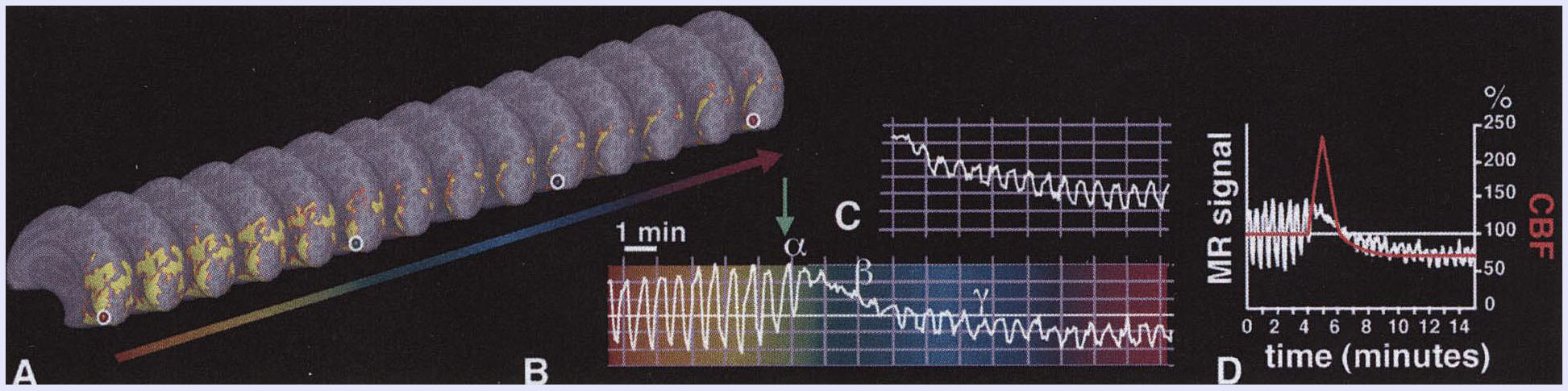
図2(Bは、図1に同じ。Cは、他の二人のうちの一人のデータ。前兆が起きてからのデータで、Bの発作の後半部分に類似しています。DはBのデータと、Lauritzenのラットの実験データと強引に合成して、時間経過がCSDに一致すると述べたものです。)
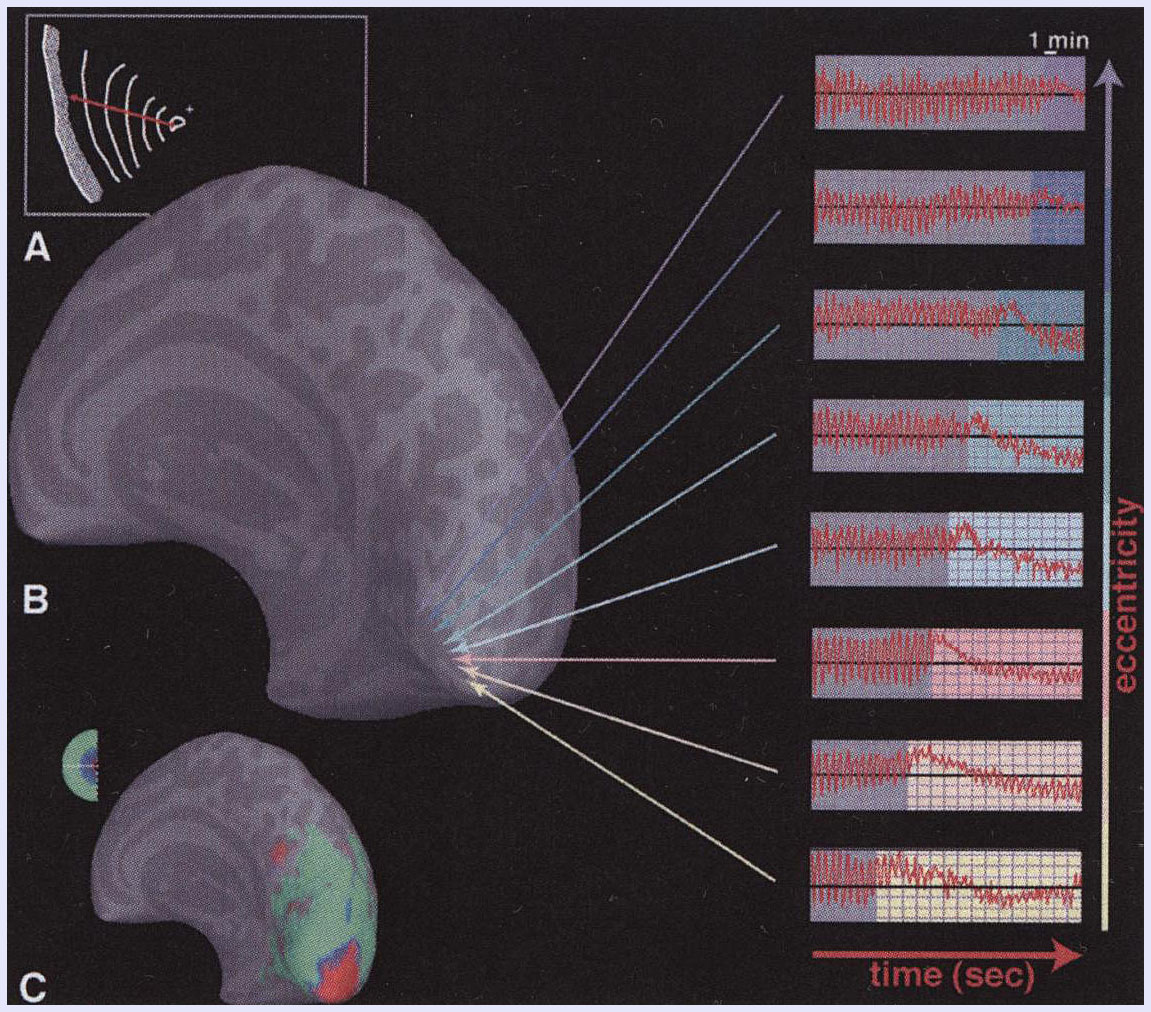
図3(図1の患者。発作前から測定が可能。(A:この患者が書いた前兆の絵。視野の異常は左側視野に生じ広がっています。C:円は、この患者の左側の視野を示しています。この色に対応したこの患者の後頭葉で視覚野。B:これらの反応は、後頭葉のV3a領域から始まり、3.5mm/分で広がっていくことを示しました。)
感想:これは、脳血管撮影で前兆のある片頭痛を起こしたOlesenらの実験結果とは段違いに信頼性が高いと思います。この報告は、2001年のものです。閃輝暗点を示す視覚性の前兆は、CSDと深い関係があると考えられていますが、現在も解明が進んでいます。しかし、未だにわからないことのほうが圧倒的に多いと思います。
CSD -part2- 【森先生の贈り物】 2016/12/12
下記に示す一番目の【CSDが三叉神経血管系の侵害受容をきたす機序について】の論文を読みながら、いろいろな論文を検索しているとCSDの論文が多数ヒットしました。
2011年1月1日に記載した1回目のCSDで記載しましたが、ここで繰り返し述べさせていただきます。
私は昭和60年に脳神経外科教室に入局しました。もう30年以上も前のことです。当時の大学の脳神経外科教室には、生理グループ、神経化学グループ、脳虚血グループの三つの研究グループがあり、私は二番目の神経化学グループに入り研究しました。生理グループチームの医局カンファランスの発表では、CSD、DC、kindling、hippocampus、CA1などの単語がよく出てきました。私は、CSDとかDCとかkindlingなどの単語がでてくるとすぐに眠くなってしまいました。当時の教室を主宰されていたのは、亡くなられた森和夫先生でした。
当時は私は神経化学班で、薬理学教室に出入りしてエンドセリンとかANPとかの受容体の研究をしていました。それが、今ではCSDの論文を検索して読んでいるとは、自分でも不思議な感覚です。
では、今冬は、CSDの論文を楽しんでみましょう、と言いたいところですが、難しいので私が好きなように読んでみます。
| Pietrobon&Moskowitz part1 | Pietrobon&Moskowitz part2 | CSD-SAH その1 | |
| Dreiner part1 |
| 著者 | タイトル | |
| 1 | Pietrobon D, Moskowitz MA | Pathophysiolgy of Migraine. Ann. Rev. Physiol.2013.75:365-91 |
| 2 | Pietrobon D, Moskowitz MA | Chaos and commotion in the wake of cortical depression and spreading depolarizations. Nature Rev. 2014.15:379-393 |
| 3 | Kramer D, Fujii T | Cortical spreadin depolarization: Pathophysiology, implication, and future directions. J Clin Neuroscience. 2016.24:22-27 |
| 5 | Dreier JP | The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease Nature medicine. 2011.17:439-447 |
2016/12/22
最近、頭痛の学会や研究会に参加するとよく呈示される論文があります。
それほど新しい論文ではないのですが、なかなか入手することができずにいました。
やっと入手することができたので紹介します。
タイトルは【Pathophysiolgy of Migraine】です。筆者はPietrobonとMoskowitzです。
出典はAnnu. Rev. Physiol. 2013.75:365-91
2013年の論文です。本来は頭痛外来をしている時に読んでおくべきでした。
その頃はいろいろな論文を読んでいたのに、検索できていなかったようです。
この論文は、片頭痛の病態生理学というタイトルですから、片頭痛の本質に迫るぞという意気込み感じられます。Moskowitz先生の研究室からの報告です。
この図に魅かれました。
論文は26ページに及ぶ大作ですが、これらの図に集約されているのではないでしょうか。
まず、この図をみてみましょう。
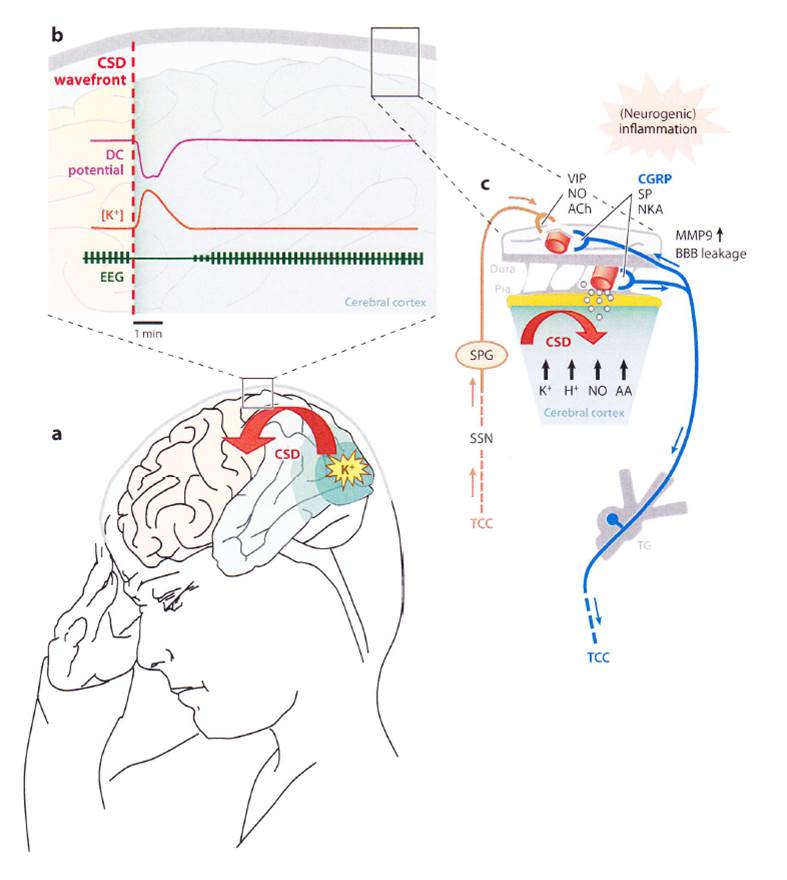 |
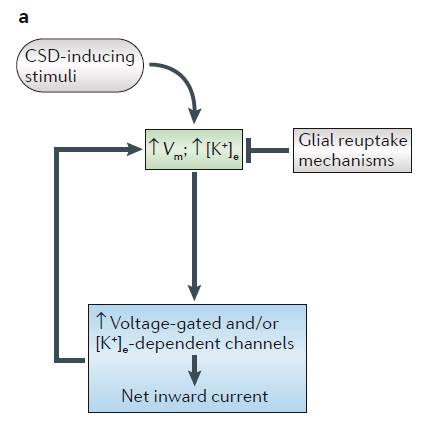
CSDが三叉神経血管系の侵害受容をきたす機序について
a)大脳皮質の神経過活動の結果、細胞外カリウムが上昇し、CSDは引き起こされると考えられています。
b)CSDは、脳波の抑制と細胞外カリウムの上昇を伴った、神経細胞とグリア細胞の脱分極がゆっくりと拡がっていく波であります。
c)CSDの間、グルタミン酸や他の神経伝達物質の他に、有害となる物質、すなわち水素イオン、NO、アラキドン酸、セロトニンなどが放出されます(図では白丸で示してあります)。
これらの物質は脳軟膜血管を支配する三叉神経血管系を刺激し、三叉神経脊髄路核複合体の中枢の三叉神経血管系の神経が賦活化されます。この一方で、側枝を介し硬膜三叉神経求心性神経線維は、脳血管関門を破壊しながら髄膜求心性神経線維に接近していきます。髄膜求心性神経線維の刺激により、CGRP、サブスタンスP、NKAなどの前炎症性血管活動性神経伝達物質が放出されます。この結果、硬膜では神経原性炎症が進行し、三叉神経血管系求心性神経線維の活性化が継続し感作が成立します。
また、CSDにより放出された物質は、感作と髄膜侵害受容の次の活性化を惹起します。
そして上唾液核と翼口蓋神経節の活性化による副交感神経系の反射は、髄膜の副交感神経遠心性線維からVIP、NO、アセチルコリンを放出します。
と、これまで報告された論文と同じような内容ですね。ただ、よくみてみると、細胞外カリウムの上昇が原因としてあるようにもみえます。神経過活動は、hyperactive neuronal circuitを訳しました、正しいのか、よくわかりません。hyperactive neuronal circuitが細胞外カリウムを上昇させてCSDを引き起こすということのようです。
この次に家族性片麻痺性片頭痛を引き起こす遺伝子を組み入れたマウスの実験にも触れてありました。全訳を行なってみましたが、意味があまりよくわかりませんでした。興奮性神経と抑制性神経のアンバランスが生じ、興奮性が増し、CSDが引き起こされる、これにはグルタミン酸、特にNMDA受容体の関与するなどの記載がありましたが、ついていけませんでした。図のみ紹介します。
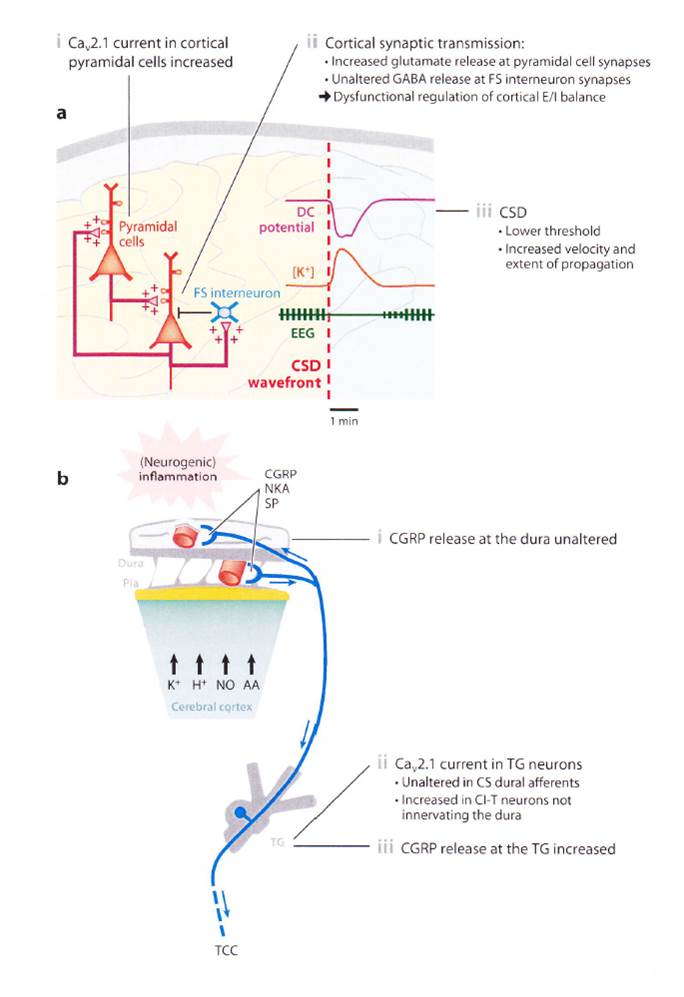 |
この論文を読んだ後に、CSDに関していくつか興味深い論文がありましたので、今後、適宜追加していきます。
Pietrobon&Moskowitz part22017/01/11
PietrobonとMoskowitzの二つ目の論文を読みながら、私が持ったイメージを付け加えていきます。
はじめに
CSDは、約1分間持続する脳神経のほぼ完全な脱分極と、引き続き数分間にわたり脳電気的活動が静まる、急激に生じるゆっくりと広がる波であります。
CSDは、イオンの恒常性の破綻、イオン勾配の崩壊、神経伝達物質のリリースからなります。
正常な代謝を有した脳組織での実験的CSDの形成は、細胞外のKイオンを上昇させる強い脱分極刺激を必要とします。
これに対して、代謝の障害された脳組織で、CSDに似たspreading depolarizationは、突然に生じるほぼ完全な神経の脱分極とイオンの再分布を示し、CSDと同様にゆっくりと広がります。
spreading depolarizationは、イオン勾配の回復、再分極、シナプス伝達、脳機能の回復が、CSDに比較して延長し、すべてではありませんが、Na-K ATPase活性が多かれ少なかれ障害されています。
例えば、spreading depolarizationは脳虚血ペナンブラで生じます。
また、anoxic depolarizationは、低酸素、脳虚血、低血糖で惹起されます。難しいですよね。どうやら、CSDとspreading depolarizationとは違うもののようです。
また、anoxic depolarizationという単語もあるようです。
あとで述べることが、できるかもしれませんが、PietrobonとMoskowitzは頭痛研究者です。CSDとspreading depolarizationとは異なるものであるという立場のようです。
これに対し、Dreier(独の脳卒中学者)は、CSDとspreading depolarizationは移行系と捉えているようです。つまり、Moskowitzらは、CSDを良性病態と捉えたい、Dreirerらは、悪性病態になりうると考えているようです、立場が異なるようです。
RCVSの病態に対して、頭痛研究者のChen SPと脳卒中研究者のDucros Aとの構図を思い出します。
この論文の中での定義です。CSDは正常代謝を有する脳で生じるものであり、ゆっくりと拡がる脱分極と引き続き脳電位活性が抑制されるものとしています。これに対して、spreading depolarizationは、脳代謝が障害された脳で生じるものであり、神経細胞・グリア細胞のNaK ATPaseが障害され、CSDと似たようなspreading depolarizationからanoxic depolarizationまでの全ての拡がって行く脱分極としています。(とてもわかりにくいですよね)
とりあえず、CSDは、脳代謝が障害されていない脳で生じ、spreading depolarizationは、脳代謝が何らかの病態で障害されている脳で生じるものと考えます。
CSDは、神経細胞死や神経細胞に対して長期持続するダメージを生じるようなことはありませんが、spreading depolarizationは、その元となる脳虚血や頭部外傷、くも膜下出血において大きなダメージを与えることがあります。
CSD 脱分極とイオン変化
CSDは1944年にブラジルのLeaoによって報告されました。ウサギの実験で、短い電気刺激を繰り返し与えると、脳表面にspreading depressionが数分間持続し、interstitial direct potentialの陰性変化が1-2分持続することを報告しています。
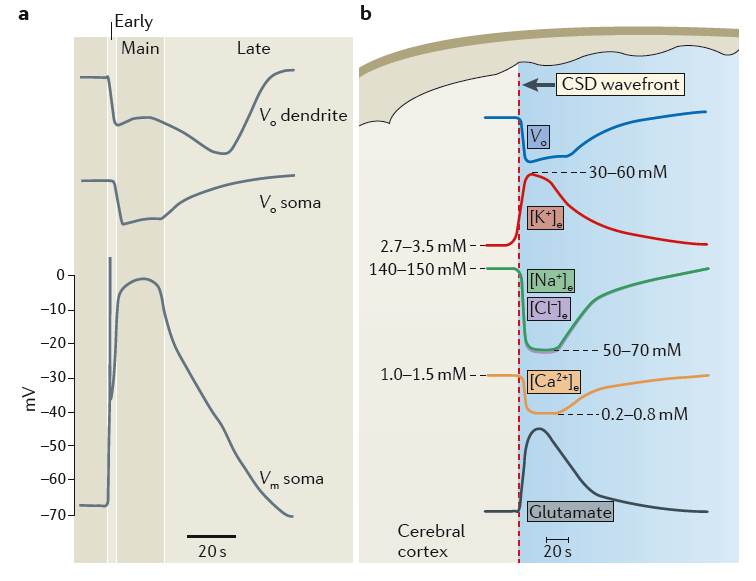 |
この図ではCSDの電気生理とイオン変化を呈示してあります。
a)の図は、stratum radiatum(放線状層)に高濃度のカリウムイオンを注入し作成されたCSDでの電気生理変化を示しています。上段は海馬における尖端樹状突起の、中段は錐体細胞の細胞体の細胞外電位(Vo)で、同時に記録したものです。下段はCSDでの、錐体細胞の膜電位を記録したものです。
Voの変化は、early peakとslow second negative peakがみられます、特にCA1の尖端樹状突起層(放線状層)で、second peakは顕著となっています。
CSDは、個々の神経細胞における脱分極の縦断勾配によって特徴つけられ、異なった相を呈する複雑な現象です。stratum radiatum(放線状層)では、stratum pyramidale(錐体細胞層)より早く脱分極が始まり、より長く持続します。
Early phaseでは、尖端樹状突起はほぼ完全に脱分極していますが、細胞体では不十分にしか脱分極していません。Early phase は数秒間です。Main phaseでは、尖端樹状突起も細胞体も完全に脱分極し15-20秒持続します。Late phaseでは、尖端樹状突起の近位部の狭い領域ではまだ脱分極が残っていますが、細胞体では、部分的に再分極しています。
言い換えてみましょう。
Early phaseでは、錐体細胞尖端樹状突起に存在するチャンネルの活性化によりCSD-related depolarizationが始まります。
Main phaseでは、引き続き細胞体や樹状突起においても他のイオンチャンネルの活性化が生じます。
Late phaseでは、somatobasal zone(細胞体基底域)でイオンチャンネルが閉鎖していきます。しかし、尖端樹状突起の近位部の狭い領域では内向き電流(net inward current)があります。
一旦、CSDが始まると、波のように隣接する灰白質を2-5mm/minの速さで拡がっていきます。
b)の図は、CSD depolarizationにおけるKイオン、Naイオン、Clイオン、Caイオン、グルタミン酸の細胞外濃度の変化を示しています。
CSDのearly phaseでの早期の細胞外電位(Vo)の変化は、細胞外カリウムイオン濃度の急激な増加(2.7-3.5mM→30-60mM)、細胞外Naイオンと細胞外Clイオンの減少(140-150mM→50-70mM)、細胞外Caイオンの減少(1.0-1.5mM→0.2-0.8mM)を伴います。
CSD depolarizationとイオンの変化は、大脳皮質を図の右から左へと広がっていくものと考えられます。赤い点線はCSDの波の前面を表しています。細胞外Kイオンの上昇は、early rapid Voの変化を伴なう細胞外Kイオンのさらなる上昇を導きます。CSDの広がりは、主に細胞外液のKイオンの拡散によるものと考えられます。
メカニズム
CSDのはじまり
 |
a) CSDのはじまりについて =モデル=
正常脳において、CSDを引き起こす刺激は、グリーンボックスで示すように、神経脱分極とKイオンの上昇を引き起こします。CSDを起動するpositive-feedback cycleには、ブルーボックスで示すように、電位依存性あるいは細胞外Kイオン依存性のチャンネルの活性化による内向き電流が必要となります。グリア細胞による細胞外Kイオンの除去が間に合わななければ、完全な神経脱分極が生じます。
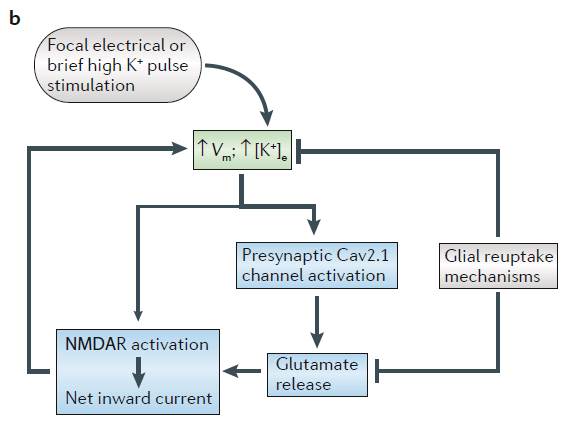 |
b) 短いKイオンパルス刺激あるいは電気刺激により引き起こされるCSDのはじまりについて
短いKイオンパルス刺激あるいは電気刺激により、皮質の錐体細胞シナプスから電位依存性Caイオンチャンネル(Cav2.1チャンネル これはP/Q-type Caイオンチャンネルとして知られています)によるグルタミン酸のリリースとNMDA受容体の活性化が生じ、CSDを推進するpositive-feedback cycleが引き起こされます。グリア細胞は、Kイオンとグルタミン酸の再吸収し、positive-feedback cycleを弱める働きを有しています。
NMDA受容体拮抗剤は、CSDをブロックします、NMDA受容体は、CSDのはじまりおよびひろがりに不可欠なものと考えられています。
まとめると、CSDは、細胞外Kイオンの増加、シナプスでグルタミン酸のリリース、NMDA受容体の活性化によってはじまります。一方、アストロサイトでは、Na-K ATP aseの働きにより、Kイオンを除去し、間接的にグルタミン酸も除去し、CSDを弱まらせる働きがあります。
ピンクリック、電気的刺激、Kイオン刺激によるCSDの薬理学的特徴は、若干異なっており、これをテーブルに示します。
| Type of depolarization | NMDAR | Cav | Nav | Zn2+i |
| CSD | ||||
| CSD1(brief K+ pulse) | ++++ | ++++ | + | no effect |
| CSD2(higf K+) | ++++ | ++ | ++ | ND |
| CSD3(pinclick) | ++++ | + | +++ | ND |
| Spreading depolarization | ||||
| anoxic depolarization | + | ++ | +++ | +++ |
| Ouabain-induced | + | ++ | +++ | +++ |
CSDのひろがりについて
CSDは典型的にはゆっくりとひろがっていきます、このゆっくりとしたスピードは、化学的物質の拡散によるものではないかと考えられています。CSDでは、Kイオンの細胞間質への拡散が重要と考えられていますが、拡散する物質や拡散する経路については未だ不明な点も多く、細胞間質への拡散、gap junctionのopening、細胞間質への液性因子、Kイオン・グルタミン酸の拡散などの問題点があります。
gap junctionについては、gap junction blockerによりCSDの拡がりが抑制されなかったことから否定的に考えられており、液性因子の拡散については、microdialysisの結果から肯定的に考えられています。Kイオンとグルタミン酸が灰白質に拡散することにより、CSDが拡がり、positive-feedback cycleがすすみます。多くの実験結果から、Kイオンがグルタミン酸よりも拡散物質として重要と考えられています。
Spreading depolarizationのはじまりについて
前述したように、この論文では、Spreading depolarizationは脳代謝が障害された状態で生じるものとしています。Spreading depolarizationにおけるイオンチャンネルやメカニズムは、hypoxia(低酸素)やOGD(oxygen-glucose deprivation 酸素-糖 貧窮状態)の後に数分間、進行するSpreading depolarizationの研究によってなされています。
anoxic depolarizationについて
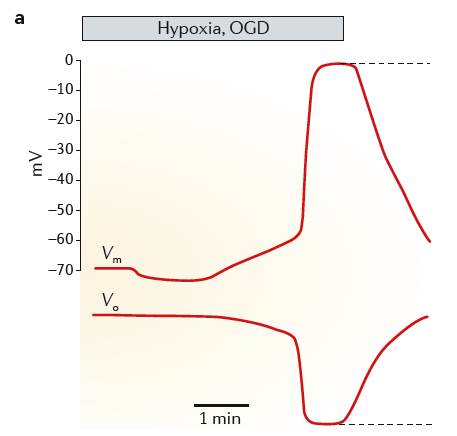 |
a)の図は、脳スライスの実験でhypoxiaやOGD(oxygen-glucose deprivation)により惹起された、典型的な膜電位(Vm)と細胞外電位(Vo)の変化を示しています。 数分間遅れて、hypoxiaやOGDはいわゆるanoxic depolarizationを引き起こします。 これは急激に生じる完全な神経の脱分極と細胞外電位の変動により特徴つけられます。 anoxic depolarization間に生じる細胞外液のイオンの変化は、CSDの変化に類似します。anoxic depolarizationの後は、組織は急速に酸素を得て、膜は再分極しイオン勾配は回復します。
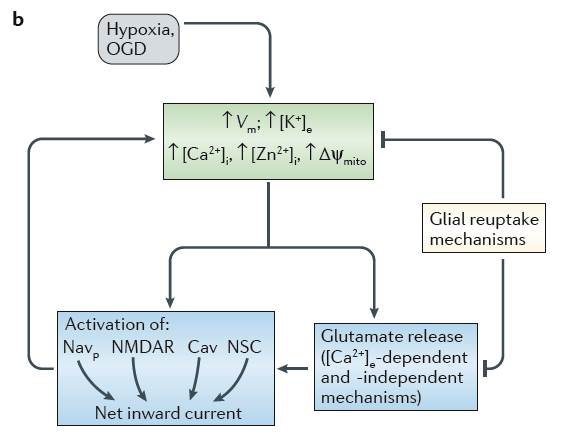 |
b)の図はanoxic depolarizationがはじまるメカニズムを示しています。
anoxic depolarizationのはじまりを推し進めるpositeve-feedback cycleの内向き電流は、多数のイオンチャンネルを介しています。
すなわち、NMDA受容体、電位依存性Naイオンチャンネル(Nav)に加えて、細胞内Caイオン増加に伴う非特異性陽イオンチャンネル、電位依存性Caイオンチャンネル、亜鉛イオンによるミトコンドリアの脱分極によって生じる因子などです。Ca依存性、非依存性のグルタミン酸リリースはanoxic depolarizationのはじまりおけるNMDA受容体活性に寄与します。ブルーボックスにある様々なイオンの寄与は、年齢や脳の部位、そして脳代謝の異常の程度によって異なります。
細かくみていきましょう。 anoxic depolarizationのはじまりは、短い過分極に引き続きゆっくりとした膜の再分極により特徴漬けられます、NaClと水との細胞内流入、pHの低下、ゆっくりとしたKイオン・Caイオン・Znイオンの上昇がみられます。この間、ミトコンドリアはゆっくりと脱分極します。 Hypoxiaの後、excitatory(興奮性)およびinhibitory(抑制性)のシナプス電位のfrequencyが増します。逆にaction potential-evoked synaptic transmissionは低下し、pre-anoxic depolarization phaseの状態まで抑制されます。 十分には解明されていませんが、NMDA受容体、Cavチャンネル、TTX(テトロドトキシン)感受性Naイオンチャンンネルなどは、内向き電流の維持に、hypoxiaの後の数分間のanoxic depolarizationの始まりに関与していると考えられています。 Caイオン非依存性および依存性のグルタミン酸リリースはNMDA受容体を活性化しanoxic depolarizationの始まりに関与しています。Caイオン非依存性の機序としてグルタミン酸はグルタミン酸トランスポーターが関与しています。 anoxic depolarizationの始まりに対して最も抑制的効果を持つのは、Navチャンネルの抑制です。TTX、リドカイン、ディブカインは全て、anoxic depolarizationの始まりに対して強力な遅延効果やブロック作用を有すると報告されています。 海馬において、anoxic depolarizationを完全にブロックする方法はNav・Cavの抑制、NMDA受容体拮抗剤、AMPA受容体拮抗剤、低温をcombinateすることです。 ouabain-induced spreading depolarizationは、hypoxiaやOGDにより惹起されたspreading depolarizationに極めて類似します。 anoxic depolarizationあるいはouabain-induced spreading depolarizationの始まる機序は、CSDの始まる機序とは明らかに異なります。 Spreading depolarizationがはじまる数分間は、特別な経過がみられます。Naイオンの流入、slow mitochondrial depolarization、Znイオンの流入などです。これらはspreading depolarizationのはじまるpositive-feedback cycleに重要ですが、正常脳におけるCSDでは働いていません。 CSDのはじまるpositive-feedback cycleにはNMDA受容体を介した機構が重要なイオンチャンネルとなります。しかし、spreading depolarizationではNMDA受容体を介した機構に加え、他のイオンチャンネル、恐らくはNavが関与していると考えられています。 spreading depolarizationのはじまりではCa非依存性・non-vesicular グルタミン酸リリースはNMDA受容体を活性化させますが、CSDのはじまりでは、Ca依存性・vesicular グルタミン酸リリースがNMDA受容体を活性化させます。
CSDおよびspreading depolarizationの脱分極の維持
CSDおよびspreading depolarizationの間、神経の脱分極を維持するものは、非選択的陽イオンチャンネルを介したNaイオンの流入によることは明らかではあるが、これらのチャンネルについてはいまだ不明な点も多い。NMDA受容体は、CSDおよびspreading depolarizationの間、脱分極の維持に寄与する。
Na-K ATP aseの薬理学的抑制を介したSpreading depolarization
Na-K ATP aseを抑制するouabainは、海馬などの実験系において数分遅れて、spreading depolarizationを引き起こします。
Ouabainにより生じたspreading depolarizationは、多くの点でanoxic depolarizationに類似していますが、正常脳におけるCSDとは全く異なっています。
実例を挙げると、Ouabainにより生じたspreading depolarizationは、Naイオンチャンネルブロッカーであるテトロドトキシンやディブカインによって抑制されます。逆にCaイオンフリーの溶液の中でもOuabainにより生じたspreading depolarizationは認められます。
Spreading depolarizationのはじまりは、細胞外液のKイオンのゆっくりとした上昇、細胞内亜鉛イオンのゆっくりとした増加、ミトコンドリア脱分極によってなされます。
CSDとmigrainous brain
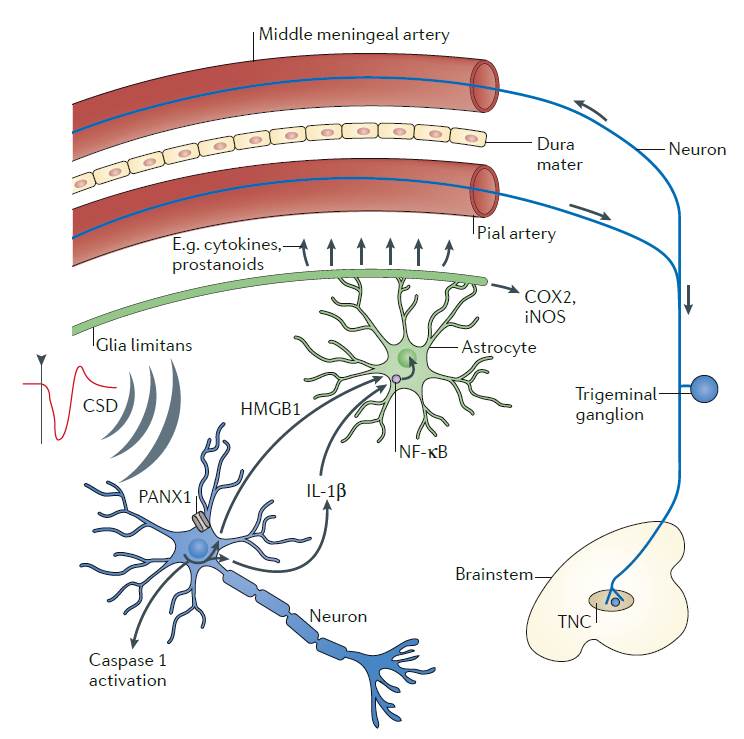 |
この図は、CSDがtrigeminovascular systemを賦活化するシステムを示しています。
筆者らの以前の論文に比べるとやや革新的となっています。
CSDが、Neuronal pannexin(PANX1)チャンネルを活性化すると、pro-inflammatory action signaling molecule (high mobility group B1 (HMGB1)、interleukin-1β(IL-1β))などがリリースされます。
その他pro-inflammatory actionとして、astrocytic nuclear factor-κβ(NF-κβ)nuclear translocationの活性化、iNOSやcyclooxygenase 2(COX2)の発現などがあり、サイトカインやプロスタグランジンなどが生成されます。
サイトカインやプロスタグランジンはグリア細胞からリリースされ、グリア細胞限界膜を通過し、脳軟膜や硬膜の三叉神経の軸索に到達します。これは、順行性に三叉神経神経節から三叉神経脊髄路尾状核に到達します。また逆行性に中硬膜動脈を刺激し、CSDによる神経原生炎症および肥満細胞からの脱顆粒を推し進めます。また、脳幹を介した反射により中硬膜動脈を拡張させます。
CSD-SAH その12017/02/12
Cortical spreading depolarization: pathophysiology, implications, and directions
私がこの論文を読みたいと思ったのは、論文で紹介されているこの図(Fig1)をみたからです。
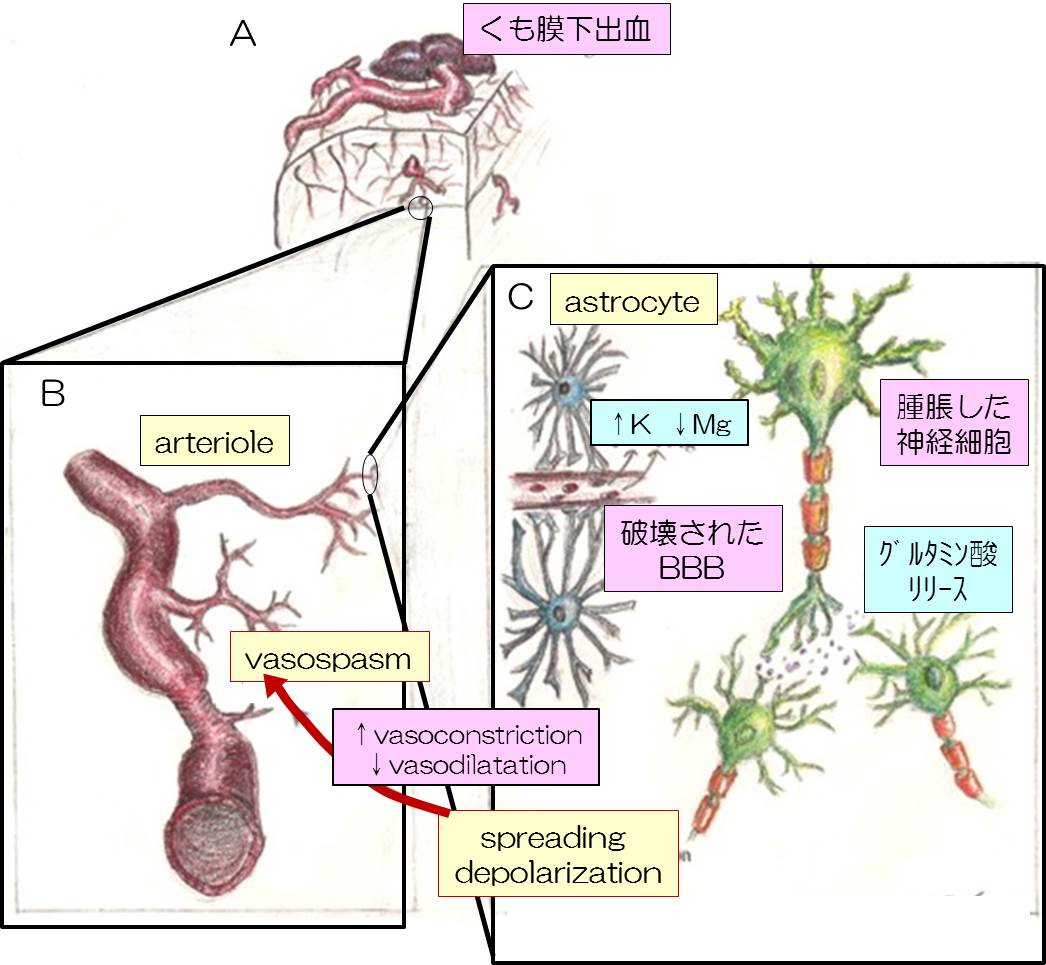 |
図の説明を、私なりに全訳してみましょう。
神経損傷後のcortical spreading depolarizationと血管反応のメカニズムについて。ここでいう神経損傷とはくも膜下出血のことです。
(A)脳動脈瘤破裂による脳損傷は、微小環境に変化をもたらします。
(B) cortical spreading depolarizationにおける、細動脈レベルの脳血管収縮と乏血反応。
(C)神経損傷による脳血液関門(BBB)の透過性の上昇は、細胞外Kイオンの上昇とMgイオンの減少により、微小環境に変化をもたらします。腫脹し、酸性に傾いた神経は、グルタミン酸と神経伝達物質をリリースし、神経周囲では興奮性毒性の状態に陥ります。このようにして、cortical spreading depolarizationは拡延し、脳血管れん縮と脳虚血をきたすという悪循環に陥ります。
奥が深すぎて、この図の説明だけではよくわかりません。脳動脈瘤破裂によるくも膜下出血により、cortical spreading depolarizationが生じ、脳血管の収縮をきたします。これにより、cortical spreading depolarizationが助長され、さらに脳血管の収縮をきたし脳虚血状態となるという負のスパイラルを示しているのだと思います。
次に本文を紹介します。この論文では、Cortical spreading depolarizationと Cortical spreading depressionとは区別されているようです。
要旨
cortical spreading depolarizationでは、イオンの恒常性の消失の拡がり、血管反応性の変化、シナプス構造の変化、elecrical activityの抑制がみられます。これに引き続いて、 neurological injuryが生じます。
この事象は、動物実験で知られていましたが、最近、人にも生じることが証明され、片頭痛や脳血管障害、てんかん、頭部外傷で生じることが知られています。
健康な脳では、cortical spreading depolarizationによりelecrical activityが増加し、NOやアラキドン酸のリリースを伴い、脳血管の拡張、脳血流量の増加、脳代謝の増加を伴います。一方、損傷した脳では、cortical spreading depolarizationにより脳血管が収縮して脳虚血をきたし、脳損傷を悪化させます。この場合、cortical spreading depolarizationが拡延し、さらなる悪化を招くサイクルに陥ります。
NMDA受容体拮抗剤やイオンチャンネルブロッカーはcortical spreading depolarizationによるダメージを減少させる効果があります。
①Introduction
1944年 Leaoは、ウサギの脳波の実験で、ある脳波の変化を発見し、cortical spreading depressionとして発表しました。イオンの恒常性の消失と脳波の抑制の拡延を認めたのです。
cortical spreading depression やcortical spreading depolarizationを伴ったcortical spreading depressionは、当初は動物実験でのみ、認められるものと考えられていましたが、人でも証明されました。脳が興奮した結果、神経とアストロサイトの機能低下が生じ、さらに脳血流量の変化を惹起します。この脱分極の波は、神経の膨張や樹状突起の変化、電位(electrical potential)の消失を伴います。
Cortical spreading depolarizationは、Kイオンやグルタミン酸により、あるいはくも膜下出血や脳外傷により出現します。
これらの侵害刺激により、神経環境が破壊され、グルタミン酸による有害事象が始まります。
グルタミン酸はCaイオンチャンネルやNaイオンチャンネルを活性化させ、特にNMDA受容体を介して陽イオンを流入させ、膜電位は-70mVから-10mVに脱分極します。
正常な状態では、この電位の変化はNa-Kポンプにより回復しますが、脳が損傷した状態ではすぐには回復しません。陽イオンの移動に伴って、神経は膨張して構造変化がみられ、膜電位が変化して脳表面の電位は抑制されます。
これが、Leaoが報告した現象です。この脱分極はおおよそ2-6mm/minのスピードで拡がっていきます。
②Vascular response
Cortical spreading depolarizationにより、脳血管の反応は変化します。
正常な神経活動では、活動電位とシナプス後電位は、グルタミン酸をリリースとNOやアラキドン酸の産生がみられ、脳血管は拡張し脳血流量は増加します。
エネルギー消費の多くは再分極のために用いられます。Cortical spreading depolarizationの間、イオンの恒常性の消失がみられ、それを回復するには多大のエネルギーを必要とします。
健康な脳組織では、CSDの後に、神経は恒常性を回復するために脳血管は拡張します。
これに対して、脳外傷や脳卒中などの病的な状態では、神経が恒常性を回復するために脳血管の拡張は得られず、脳虚血に陥ります。
Cortical depressionの間、エネルギー需要は増加し、グルコースは低下し乳酸が増加します。NOやアラキドン酸などの物質は、恒常性回復のため必要なエネルギーを提供すべく血管拡張反応を示します、これは正常な神経活動に類似します。
しかし、cortical spreading depolarizationでは、周囲の微小環境では、酸性、高Kとなり脳血管収縮をきたします。NO利用の低下、高K、酸性などは、異常なoligemia(乏血)状態となります。
拡延するhyperemia(充血)は2分間ほど続き、引き続きoligemia(乏血)となります。oligemiaは、血管反応性の低下のためneurovascular couplingをともなっていません。
境界領域では、エネルギー需要の増加に脳血流増加が伴っていないため相対的に脳虚血に陥ります。貧窮な灌流は、新たなoligemiaがcortical spreading depolarizationの影響を受けやすい部位を形成するという悪循環をきたし、結果的に脳虚血が拡がっていきます。
③Etiology of cortical spreading depolarization cortical spreading depolarizationの原因には、神経の脱分極を起こす化学的刺激、機械刺激、電気生理学的刺激があります。このような刺激とcortical spreading depolarizationとの結びつきの可能性として、脳血流関門のオープニングがあげられます。 BBBの透過性が増し、Mgイオンが下降すると、NMDA受容体を介して脱分極が増加し、細胞外Kが増加します。これらがcortical spreading depolarizationのはじまりと関係しています。アルブミン流入に伴い、Kのクリアランスが低下することも、このカスケードを助長します。 その後、neuronのいたるところで脱分極は広がり、neuronの腫脹、酸性の環境、過分極の消失が拡がっていきます。 腫脹し酸性となったneuronは、グルタミン酸や他の神経伝達物質をリリースし、神経周囲に興奮性毒性を生じる。このようにして拡延はさらに広がっていきます。周囲のダメージを受けた組織は、脱分極や過興奮の影響を受けやすくなっています。 異常な血管反応により、低灌流は周囲に広がり、流入した陽イオンを処理するためのエネルギーを供給できなくなります。この危険なサイクルは、脱分極の領域では継続します。このサイクルは、脱分極の波が、恒常性を回復することができる正常組織に到達すると停止します。
④ cortical spreading depolarization in humans
cortical spreading depolarizationは、動物実験では確認されていましたが、人でも生じていることが証明されました。
そして、cortical spreading depolarizationの40-70%に脳波でspreading depresionが認められています。
⑤⑥⑦⑧ 外傷性脳損傷 脳卒中 くも膜下出血 てんかん
cortical spreading depolarizationは、外傷性脳損傷では50-70%、脳卒中では35-88%、くも膜下出血では70%にみられると報告されています。てんかんとcortical spreading depolarizationについては、その関係はまだ混とんとしているようです。
研究が進んでいる、cortical spreading depolarizationとくも膜下出血についてみてみましょう。
脳動脈瘤破裂によるくも膜下出血で、cortical spreading depolarizationは、前述のoligemiaが問題となります。
脳動脈瘤破裂によるくも膜下出血の予後には、手術などで脳動脈瘤の治療が上手くいっても、その後のdelayed cerebral ischemia(DCI)(遅発性脳虚血)が問題になっていました。
DCIの原因として脳血管れん縮(vasospasm)が考えられてきました。しかし、脳血管れん縮が画像で明らかとなるのは、DCIの50%にしかすぎません。
DCIとcortical spreading depolarizationの微小環境は類似しています。Kの上昇、NOの低下、すなわち脳血管収縮が導かれ、脳虚血に陥るということです。
脳血管れん縮がなく、cortical spreading depolarizationによるDCIも術後のモニタリングで証明されたものもあります。
cortical spreading depolarizationでは、大きな動脈の血管収縮をきたすことはなく、むしろ細動脈レベルの血管攣縮をきたします。細動脈レベルでの脳血管収縮は、脳虚血をきたすばかりでなく、脳血管撮影ではとらえられないという特徴をもちます。
cortical spreading depolarizationは、DCIによる付帯現象である可能性もありますが、cortical spreading depolarizationでみられる脳血管の反応は、DCIのメカニズムを考える上で魅力的で、DCIの重要な治療手段が発見される可能性もあります。
⑨Pharmacological targeting
NMDA受容体拮抗剤やCGRP受容体拮抗剤などあげ、治療の可能性を模索しています。
Dreiner
part12017/05/15
abstract
spreading depolarizationは、灰白質の波です。ニューロンの腫脹、樹状突起のねじれ、slow electrical potentialの大きな変化、brain electrical activityのsilencing (spreading depression)などの特徴を有します。
現在では、臨床の場面で、人でspreading depolarizationが、くも膜下出血、脳虚血、脳外傷などで生じる事が確認されています。
実験では、カリウム、グルタミン酸、Naポンプ拮抗剤、てんかん、低血糖、虚血などでspreading depolarizationを作成しています。
spreading depolarizationに反応し、正常状態では脳血管は拡張し、脳虚血などの病的状態では収縮します。
spreading depolarizationをターゲットにして、これらを引き起こす疾患を加療できる可能性が検討されています。
mechanism of spreading depolarization
spreading depolarizationの主な作用部位は、ニューロン及びその樹状突起近位部です。
ニューロンは神経シグナルを伝える電気化学的力を蓄えていると考えられています。膜を介するイオン勾配、電気チャージには、Na、K、Clが重要な役割を果たしています。イオン勾配のホメオスタシスを保つためにはNaポンプが必要です。脳は、全エネルギーの20%を消費し、その半分をNaポンプが消費します。その為、脳は生体の中でも虚弱性が高い部位と考えられます。
spreading depolarizationは、ある意味では、脳のホメオスタシスの機能停止状態といえます。イオン勾配の維持がほぼ完全に機能停止した状態で、ニューロンの脱分極の持続、ニューロンの腫脹、樹状突起の捻れなどが特徴的です。
ニューロンの細胞内外で、short circuitがみられ、生理的、形態的にニューロンのcytotoxic edemaが生じています。
spreadeing depolarizationは脳の電気静止状態を形成し、これがspreadeing depressionと呼ばれています。spreadeing depolarizationでは、細胞外スペースでは、slow potentialの陰性波がみられます。このslow potential changeはニューロンに沿った脱分極により形成され、樹状突起の層でイオンチャンネルが開きNaやCaが流入するためと考えられています。
拡がるということは、spreadeing depolarizationの本質ではありませんが、spreadeing depolarizationの核となる進行のスピードは2-6mm/minとされています。
ニューロンとニューロンのgap junctionによるコミュニケーションが、spreadeing depolarizationが拡がる際に細胞を同時に作用させ、細胞間連絡路として機能していると考えられています。この場合、ニューロンの興奮は必要ありません。もちろん、アストロサイトの機能にも関係なく拡がっていきます。
spreadeing depolarizationに際し、グルタミン酸、アセチルコリン、GABAなどがリリースされ、グルタミン酸はイオンチャンネルを開き、陽イオンを流入させ脱分極を維持します。
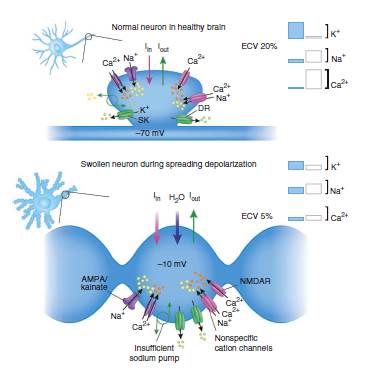 |
the normal hemodynamics response to spreading depolarization
neurovascular couplingにより、神経活動に伴い脳血流量は上昇し、神経活動の低下に伴い脳血流量は低下します。
神経興奮に伴い、シナプス後神経では、グルタミン酸によりCaイオンが流入しNOやアラキドン酸の産生が亢進します。
この血管拡張は、シナプス前の活性及びシナプス後の脱分極を反映します。シナプス後の脱分極はNMDA受容体で、Mgによるブロックを取り除かれ Caが流入します。
アストロサイトは、代謝性グルタミン酸受容体を介して血管平滑筋に情報伝達します。
spreadeing depolarizationは、生理学的に神経を活性化し、さまざまな特徴を示します。グルタミン酸やNOなどの血管拡張物質をリリースし、脳血管は拡張します。脳代謝は亢進しエネルギー需要は高まり、NaやCaイオンのポンプ機能も高まります。
即ち、spreadeing depolarizationに反応して脳血流量は増加します。
これは、spreadeing depolarizationに対する正常なneurovascular couplingの反応です。しかし、spreadeing depolarizationに際しては、神経伝達は抑制されます。
脳血流量増加は、spreadeing hypermiaと呼ばれます。spreadeing hypermiaは2分間持続し、その後は、血流は低下します。この血流低下は、spreadeing oligemiaと呼ばれています。
 |
spreading depolarization indicates brain pathology
spreadeing depolarizationの生理的役割は不明です。健康な正常な組織では、普通の状態では生じません。spreadeing depolarizationの電気的、イオンや代謝の異常は、てんかん発作時の異常より大きくなっています。
spreadeing depolarizationは、有害刺激、機械的損傷、電気的刺激、低浸透圧、高温、Kイオン、グルタミン酸やアセチルコリン、急激な過興奮状態、てんかん重責、Naポンプ阻害剤、低血糖、低酸素、脳虚血により生じます。
有害刺激にあった部位から、spreadeing depolarizationは正常組織に拡がっていきます。細胞下の進行は有害刺激の性状によりますが、このnonlinear waveの本質的生理学的な特徴は、有害刺激の高い所から低い所に沿って拡散する間は維持されます。
局所の回復はNaポンプ活性の再生によります、これは有害刺激のもと、障害されています。
再分極のないところでは、spreadeing depolarizationはterminal spreadeing depolarizationとよばれます。terminal spreading depolarizationは多源性であり、重度の有害刺激を伴っています。低酸素、低血糖、心停止、高K・グルタミン酸・ウアバイン・Naポンプインヒビターなどによります。
有害刺激が中等度のものでは、spreading depolarizationは再発する一塊を形成し、中等度の期間です。
有害刺激が軽度のものではspreading depolarizationは単一で短時間です。これは健康な脳で生じ、直ぐに(1分程度)回復します。
terminal spreading depolarizationは通常neuronal death(神経細胞死)を伴ない、短時間のspreading depolarizationでは、神経細胞は生存します。
本論文は、神経細胞が生き残るのか死滅してしまうのかを見極めることを目標としています。Spreading depolarizationがある時間まで延長すると細胞内のシグナルが始まり、神経細胞は死滅します。この時間はcommitment pointと呼ばれています。
commitment pointは、神経解剖構築、数、有害刺激の条件などにより異なります。
prolonged spreading depolarizationが細胞死を起こす機序として、細胞内Caイオンの増加が考えられています。
細胞内Caイオンは25μMまで上昇しますが、これはshort lasting depolarizationでもterminal spreading depolarizationでも認められます。
脳のスライスの実験において、低酸素でみられるspreading depolarization は、Caイオンフリーの実験系で、Caイオンが正常の実験系に比較して、シナプス伝導のより良い回復が認められます。
さらに、イオン恒常性の破綻はグルタミンの逆輸送主要な原因となります。
この事は、興奮性毒性とspreading depolarizationとが多くの点でオーバーラップすることを示しています。
The pattern of brain activity depression functional deficit
spreading depolarizationとspreading depressionを同義語として用いるべきではありません。spreading depressionはspreading depolarizationの副現象(epiphenomenon)です。spreading depolarizationは、脳電気活性のspreading depressionもnonspreading depressionも伴います。
spreading depressionはmigraineの前兆に似ています。
migraineの前兆は、creeping fashionとして出現します。典型的には閃輝暗点として両眼の一側の視野に出現します。
nonspreading depressionは、脳虚血および低酸素により突然出現します。動物実験では、脳血流が20-25ml/100g・min以下でみられます。
spreading depolarizationは、spreading depolarizationが始まる前に長い時間、脳の電気活性が停止していた場合には、spreading depressionを伴うことはありません。
spreading depolarizationが始まるまでに、脳代謝がおおまかに正常で十分なエネルギー供給がなされている場合、spreading depolarizationはspreading depressionを伴います。
このことは、突然に神経脱落症状を引き起こすことと対照的に、spreading depressionによる片頭痛の前兆の症状が、通常は予後良好であるかを示しています。
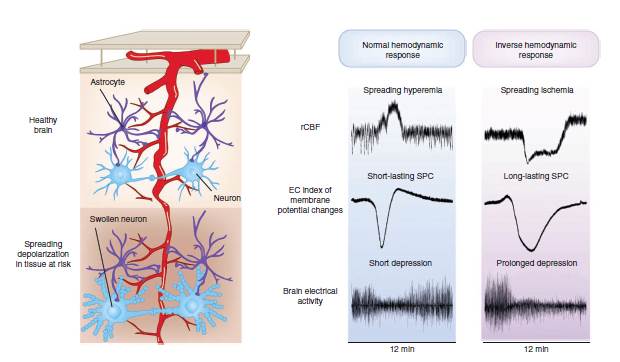
The inverse hemodynamic response to spreading depolarization
spreading depressionを伴っているにもかかわらず、ある条件下では、spreading depolarizationは、神経損傷を伴っていることがあります。
spreading depolarizationに対する血行動態反応が逆転している場合に、その可能性があります。
逆の血行動態反応とは、病的状態におけるspreading depolarizationに伴うsevere arteriolar constrictionによる著明な延長した低灌流状態のことです。
このプロセスはspreading depolarizationの間、虚血による細胞死を引き起こすことになり、spreading depolarizationの開始とともに灌流障害と代謝障害が始まっています。
spreading depolarizationにより引き起された低灌流は、spreading depolarizationからエネルギー回復が全く追いつかないほどシビアである。逆の血行動態反応(the inverse hemodynamic response)は、居所的には無害のshort-lasting depolarizationを有害のものに変化させ、terminal depolarizationにまで変化させます。
シビアなspreading ischemiaは拡がって神経細胞やアストロサイトなどの広範な皮質汎壊死につながります。しかし、spreading ischemiaを正常のspreading oligemiaと混同してはいけません、spreading oligemiaは組織再分極にともなうmildな脳血流量の低下を意味するからです。
最新の実験結果から、spreading ischemiaは微小血管反応性(microvascular reactivity)の障害により引き起こされた悪循環の結果、生じると考えられています。
spreading depolarizationは、細胞外Kの急激な上昇と著明なpHの低下によるものを伴っています。
20nM以上の細胞外Kは強力な血管収縮を惹起します、一方、pHの低下は血管拡張を引き起こします。正常な状態では、Kによる血管収縮反応に対抗するNOの強力な効果はpH下降にともなう血管拡張を助長します。よって、spreading depolarizationの効果は、通常は血管拡張です。
実験的には、わずかなKの上昇とNO利用の低下といった条件下では、spreading hyperemiaからspreading ischemiaへのシフトします。NOの利用低下という条件下では、spreading depolarizationに際し細胞外イオン変化は、血管拡張から血管収縮に変化します。さらにKの基礎値の上昇は、アストロサイトや血管平滑筋細胞で細胞内Caを上昇させ、spreading depolarizationの間、Caがリリースされ血管収縮反応が増します。
これらの因子は、spreading depolarizationの効果をvasodilatationからvasoconstrictionに変化させ、神経への酸素供給を減少させエネルギー需要を増加させます、その結果、NaポンプやCaポンプの機能低下が引き起こされます。
一方、spreading depolarizationと血管収縮物質の維持は、spreading depolarizationが血管収縮を維持し、虚血・血管収縮はspreading depolarizationを維持します。
Migrainous stroke and inverse hemodynamic response
片頭痛は女性の15-25%に認められます。片頭痛の前兆は通常は無害ですが、稀にmigrainous strokeを生じることがあります。
Migrainous strokeの定義から考慮すると、migrainous strokeはspreading depressionにより開始しており、spreading depolarizationが出現するまでは組織エネルギーの状態や代謝は正常と考えられます。
さまざまな病理生理学的現象が知られている中で、spreading ischemiaにfitするのはmigrainous strokeと考えられます。
しかし、migrainous strokeは稀な病態であり、spreading hyperemiaからspreading ischemiaにスイッチする因子が何であるのかは、あまりわかっていません。
Spreading ischemia and delayed cerebral ischemia after aSAH
実験的には、NO利用の減少と細胞外Kの上昇がspreading hyperemiaからspreading ischemiaにシフトすることが証明されています。これは、脳動脈瘤破裂後の遅発性脳虚血で認められます。すわなち、くも膜下出血の血塊のHbによるNOスキャベンジ、内因性NO合成酵素阻害、血管周囲のNO神経の変性などが証明されています。
また、くも膜下出血の血塊によるKのリリース、Naポンプの障害、microembolus、ジギタリス様物質などもK上昇に寄与しています。これらにより、spreading ischemiaがくも膜下出血後の遅発性脳虚血に関与していると考えられています。
予防治療にL型Caチャンネル拮抗剤のニモジピンが用いられています。
最近、spreading ischemiaがくも膜下出血の症例で次々と証明されています。Spreading depolarizationに対するinverse hemodynamic response が、新たな治療のターゲットとなる可能性を秘めています。
spreading ischemia inte ischemic penumbra
spreaqding ischemiaはMCA閉塞モデルのischemic penumbraに一致することが観察されています。Penumbraでは病変が拡大することが知られています。
2次元の脳血流検査より、重症の脳虚血は、逆の血行動態反応によるspreading depolarizationにより拡大していきます。逆の血行動態反応は徐々に正常のhyperemic responseの領域にも伸びていき、spreading depolarizationはpenumbraの領域から周囲の正常領域まで拡がっていきます。
ischemi penumbraにおいてもNOの利用低下、Kの上昇が議論されていますが、これはまだ更なる研究が必要と考えられます。
Spreading depolarization from bench to bedside
10倍も微小なてんかん波は脳波で描出できるのに、Spreading depolarization を示すSlow potential dischargeは、通常の脳波ではとらえることができません。
このため、数十年も間、人の脳ではspreading depolarizationは生じないと結論されてきました。しかし、硬膜下腔に電極を挿入することにより、人の脳においても明らかにspreading depolarizationが出現することが証明されました。
1970年代に尾状核と海馬で記録され、以降、いろいろな方法で証明され、Mayevskyらは脳外傷でspreading depolarizationを証明し、Strongらは、硬膜下腔に電極を挿入するという確固たる方法で、脳外傷において約50%の症例にspreading depolarizationが出現することを証明しました。
以降は、さまざまな疾患でspreading depolarizationが証明されています。くも膜下出血後の遅発性脳虚血、重症の脳梗塞、てんかんなどです。
spreading depolarizationは、くも膜下出血では72%にみられ、重症脳梗塞では82%に認められたという報告もあります。そしてNMDA受容体拮抗剤がspreading depolarizationに対し効果をあげたという報告があります。このように近い将来、spreading depolarizationが治療のターゲットとなる可能性が考えられています。
Box 1. Definition of terms
Spreading depolarization: generic term for the whole spectrum of waves in the central nervous system characterized by abrupt, near-complete sustained depolarization of neurons, observed as a large change of the slow potential.
Slow potential change: cortical neurons have a spatial orientation perpendicular to the surface. Sustained depolarization changes along the neuronal main axis. This creates a slowly changing intracellular electrical potential difference associated with a slow extracellular field potential change.
Thus, synchronous sustained depolarization of thousands or millions of neurons produces a slow extracellular field potential change as a summary measure for spreading depolarization . This slow field potential change can also be recorded using electrodes on the brain surface
Terminal depolarization: spreading depolarization without neuronal repolarization; occurs in the presence of severely noxious conditions such as anoxia, aglycemia or severe focal or global ischemia .
Intermediate depolarization: prolonged spreading depolarization with a character between terminal depolarization and short-lasting spreading depolarization . Intermediate spreading depolarizations can occur, for example, under hypoxia, hypoglycemia or in the ischemic penumbra where they are often referred to as peri-infarct depolarizations. They usually occur in a cluster of recurrent events, often ride on an ultraslow potential and are associated with persistent depression of activity between the events
Brain electrical activity: spontaneous electrical activity produced by the firing of cortical neurons. The firing of upstream neurons causes intracellular postsynaptic potentials in downstream neurons. Those postsynaptic potentials are associated with rapid extracellular field potential changes. Synchronous activity from thousands or millions of neurons with similar spatial orientation produces fast extracellular field potential changes that are recorded as brain electrical activity .
Nonspreading depression of brain electrical activity: electrical silence of brain electrical activity that develops simultaneously in a large zone exposed to severe hypoxia or ischemia within seconds8,45; precedes spreading depolarization by some minutes . Likely pathophysiological correlate of the clinical symptoms of nonmigrainous stroke and hypoxia. For example, in a transient ischemic attack the perfusion deficit triggers nonspreading depression within seconds, followed by spreading depolarization after some minutes; rapid re-establishment of perfusion entails rapid recovery from spreading depolarization without tissue damage.
Spreading depression of brain electrical activity: electrical silence of brain electrical activity as a consequence or epiphenomenon of spreading depolarization due to a depolarization block of neuronal activity .
It is likely that there are additional mechanisms of silencing the activity than the depolarization block, as the depression of activity typically outlasts the depolarization. Spreading depression propagates in the tissue together with spreading depolarization. Spreading depression can only occur if energy status and metabolism are largely intact until spreading depolarization onset. Likely pathophysiological correlate of migraine aura or migrainous stroke . Of note, experimental and preliminary clinical evidence suggests that both spreading depression and nonspreading depression can be associated in principle with either short-lasting or terminal depolarization, that is, either with
or without tissue death.
Spreading hyperemia: increase of regional cerebral blood flow in response to spreading depolarization .
Spreading oligemia: mild decrease of regional cerebral blood flow after recovery from the tissue depolarization .
Spreading ischemia: spreading depolarization induced severe spreading hypoperfusion that leads to a prolonged slow potential change .
CSD
-stroke2018-2018/03/09
CSDに関して論文を読んできましたが、さっぱりわからないという点が多く、何かしらの学会発表を聴きたいなあと思っていたら、ピッタリの学会発表(Stroke 2018 片頭痛と脳卒中の未来予想図)があるようです。
地元でもあり、早速、出席の予定としました。
一寸頭を整理して、抄録を少し覗いてみましょう。
私にとって、CSDは、Cotical Spreading Depressionの略です。ここで、CSD part1(古典的なもの)から、現在の私のCSDについての理解を箇条書きにしてみましょう。
①CSDは、実験的に細胞外Kイオンの上昇や、針刺激により誘発される。
②脱分極をきたし、その後、抑制される。これが2-6mm/minのスピードで拡がる
③②は、片頭痛の前兆の眼症状(閃輝暗点)に一致する
④脳血流は一過性に上昇し、その後、低下する、これはoligemiaと呼ばれている。
次に、CSD part2から、よくわからない点を挙げてみます。
①cortical spreading depolarizationとcortical spreadin depressionの相違について
②細胞外Kイオンの上昇がCSDの原因のようですが、細胞外Kイオンはどうして上昇するのか
③anoxic depolarizationとは
④terminal spreading depolarizationとは
⑤CSDの片頭痛、てんかん、脳虚血、くも膜下出血、頭部外傷における役割
⑥CSDとRCVSの関係について
などです。
理解できるかどうかわかりませんが楽しみです。
3題の発表があるようです。これにてんかん部門からの発表があればよいのですが、そこまではないようです。
| 演者:所属 | タイトル | |
| 1 | 杉本至健、 鈴木倫保、他: 山口大学 脳神経外科 |
くも膜下出血とCortical Spreading Depolarization(CSD) |
| 2 | 飯塚高浩: 北里大学 神経内科 |
家族性片麻痺性片頭痛(FHM)と皮質拡延性抑制Cortical Spreading Depression(CSD) |
| 3 | 畝川美悠紀、 鈴木則宏: 慶応大学 神経内科 |
大脳皮質拡延性抑/脱分極(cerebral spreading depression/depolarization(CSD)が結ぶ片頭痛と脳卒中 |
各々の演題でCSDと表記されていますが、各々異なるようです。
演題1は、Cortical Spreading Depolarization、演題2はCortical Spreading Depressionで、演題3はcerebral spreading depression/depolarizationとなっています。異なったものを発表しているのでしょうか。
-発表をきいて-
有意義でした。ひとりで思い込んでいたものが、なんとなくぼんやりとわかってきたような気もしますし、わからなかったような気もします。2018年3月15日は病棟をひと廻りしてから、参加しました。
とりあえず、3演題を振り返ってみます。なにぶん、素人なのでわかる範囲で。
杉本先生と岡先生の御発表:くも膜下出血(SAH)の手術時にモニタリングとしてプローベを挿入。1㎝感覚に6か所のプラチナ電極があり、そこからモニタリング。DC波形とAC波形により広帯域の脳波、頭蓋内圧などが測定しているそうです。
DCの変化で、spreading depolarizationが捉えられ、引き続き、ACの変化でspreading depressionが捉えられているようです。ベッドサイドで一目でみれるので簡単。br/>
SAHごのDCI(delayed cerebral ischemia)が出現した15例では全てでCSD(cotical spreading depolarization)が認められた。cluster CSD(3時間に3個以上といわれていた?)やisoelectric SD(通常はspreading depressionがすぐに回復するが、その回復が悪いもの)が悪いように発表されていた。その他に温度、NIRSなどを測定できるようにしているとのことであった。
CSDをモニタリングすることにより、DCIの出現の指標になるが、治療に結びつくか否かは今後の課題というようであった。シロスタゾール、NMDA受容体拮抗剤(ケタミンは無効?)、ラモトリギン、ガバペンチンなどの名前が挙がった。
飯塚の御発表:FHMの家系で、cortical spreading depression(CSD)を観察した報告です。19時間を目安にそれ以前では、脳血流量と脳代謝が低下し、それ以降では増加するという内容でした。
脳血流量の低下は多巣性のCSDによるものと考えらています。脳血流量の増加は、CSDから三叉神経血管系が賦活化されたもの、シナプス、アストロサイトが関与、Hamel先生の2006年の図が引用されて説明されていました。
畝川先生の御発表:cortical spreading depression/depolarization (CSD)は神経細胞やグリア細胞が一斉に興奮し、その後に持続的に抑制が周囲に伝播していく現象である。KCL局所投与後にDCとCBFを測定。
脳実質が局所的に低酸素に陥り、DCの一過性低下、脳表軟膜動脈や実質内穿通枝が著しく収縮、その後拡張するということを示されました。
脳表軟膜静脈や毛細血管には変化なし。2回目以降では、脳血流の低下はなく増加のみが認められたようです。
FHMマウスでは、KCLに対する閾値が低く、CSDを起こしやすい、伝播速度が早い、脱分極からの回復が遅いなどが示されました。
MCAOモデルのラット(田村の方法をmodified)では、CSDがみられるもの、みられないものがあり、前者は脳梗塞をおこし、後者は脳梗塞がおきなかったということです。
ニトログリセリンと頭痛2010/11/01
私の趣味は山歩きですが、今年はザイデングラードと黒戸尾根を歩きました。歩きながら頭痛のことを考えることがあります。随分昔の話ですが、医師になって三年目の頃、叔父から頭痛がすると電話がありました。よく聞いてみると、循環器内科の医師から狭心症と診断され、ニトログリセリンが処方されたようです。ニトログリセリンの副作用に頭痛があります。山道を歩きながら、そんなことを思い出していました。
 |
| 雨の黒戸尾根9合目 |
ニトログリセリンは、血管平滑筋細胞の一酸化窒素(NO)を介し、cGMPを生成し、細胞内カルシウムイオンを減少させて、血管平滑筋を弛緩させます。難しい話ですが、”ニトログリセリンは、何かしらの物質(NO)を介して心臓の血管を拡げて心臓をよくする薬剤”ということです。
ニトログリセリンは、興味深い薬剤です。1846年にイタリア人のAscanio Sobreroによって生成されました。彼は、この物質は舐めると少し甘くて、すぐに頭痛を起こすということを当時、既に記載しています。さらにこの物質は、爆発しやすく爆薬にするには危険と考えました。その後、ノーベル(Alfred Nobel)が創意工夫し、この物質を爆薬【ダイナマイト】として開発しています。彼は、ダイナマイトで、巨富を得ましたが、産業ばかりでなく軍事にも使用されたため死の商人と呼ばれ、辛い人生を送ったようです。彼は、その莫大な遺産をノーベル賞設立にあて、人類の平和と進歩に多大な貢献をしています。一方で、ノーベルは、狭心症を患い、虚血性心疾患の薬剤として開発されたニトログリセリンに助けられたとの話も伝えられています。
さて、ニトログリセリンの頭痛の話にもどります。2009年にPeer C. Tfelt-Hansenらが発表した論文【Nirtoglycerin Headache and Nitroglycerin-Induced Primary Headaches】を元にニトログリセリンの頭痛について考えたいと思います。
ダイナマイトを使う労働者たちが、休みの翌日、つまり、月曜日になるときまって頭痛を訴えていました。労働を続けていると頭痛はなくなり、決まって休み明けの月曜日の仕事で頭痛がするのです、これはMonday headacheと呼ばれました。今では、このことは、月曜日に仕事を始めて、ダイナマイトからニトログリセリンを吸収し頭痛がするようになり、時間とともに耐性(慣れ)が生じ、痛まなくなるという具合に理解されます。現在の臨床の場では、「狭心症のためニトログリセリン(類似薬剤)を用いると当初は頭痛がすることがありますが、数日間続けていくうちにあまりしなくなります」と、説明されます。・・・・ニトログリセリンによる頭痛は、ただ、それだけでしょうか。
ダイナマイトを使う労働者たちは、休日にも極めて少量のダイナマイトを皮膚に塗るという工夫により、耐性(慣れ)を継続させ、Monday headacheから開放されました。しかし、2-3%の労働者は頭痛から解放されることなく、転職を余儀なくされています。このことは、ニトログリセリンによる頭痛が、単一ではなく、他の頭痛が潜んでいる可能性を示唆しています。
Peer C. Tfelt-Hansenらの論文では、ニトログリセリン頭痛とニトログリセリンによってひきおこされる頭痛に分けてあります。ニトログリセリン頭痛は、多くの労働者たちが経験した頭痛です。ニトログリセリンを吸収し、心臓の血管が拡張するように、脳や頭蓋外の血管も拡張し、そのために頭痛が惹起されます。ニトログリセリン舌下後、数分程度で出現し、比較的短時間で消失するものです。一方、ニトログリセリンによってひきおこされる頭痛は、ニトログリセリン頭痛とは異なり、ニトログリセリン舌下後、1~数時間してから出現し、持続時間も長いという特徴があります。このタイプの頭痛は、ニトログリセリンにより、片頭痛や群発頭痛、緊張性頭痛が惹起されるものと考えられています。Petersらは片頭痛の患者にニトログリセリンを投与し71%に片頭痛が惹起されたと報告しています。ニトログリセリンで誘発される片頭痛の研究はすすみ、血管拡張とはあまり関係なく、NO-cGMPを介したもの、脳幹への刺激などが考えられています。ダイナマイトを使う労働者を頭痛で苦しめ、虚血性心疾患の患者を頭痛という副作用で苦しめたニトログリセリンは、現代の頭痛研究において重要な位置を占め、新たな治療薬の開発などにも繋がっています。
若い人でも慢性硬膜下血腫は生じる?2010/06/01
慢性硬膜下血腫の手術は、多くの脳神経外科医師が執刀医となる最初の手術だと思います。比較的容易に診断できて、手術も脳神経外科の手術で最も安全にしかも短時間でできるものです。入院期間も一週間程度ですみます。
慢性硬膜下血腫は、私が脳神経外科医師になって最初の研究テーマでした。当時約270例のデータベースを作成し、慢性硬膜下血腫の病態を稀な病態も含めて、ほとんど全て網羅していました。
慢性硬膜下血腫は高齢の人に生じやすい病態です。脳と頭蓋骨の間には硬膜という硬い膜があります。高齢になって脳が萎縮すると、脳と硬膜の間に隙間ができて、外傷などを契機として、脳と硬膜の間の隙間(硬膜下腔)に血液がたまり、慢性硬膜下血腫を形成することがあります。
では、若い人では、慢性硬膜下血腫は起きないのでしょうか。40代以下の人にも生じます。私の経験では17歳の人にもおきました。40代以下の慢性硬膜下血腫の患者様には注意が必要です。思わぬ基礎疾患が潜んでいる危険性があるからです。出血性疾患の合併の危険性も考えられます。慢性硬膜下血腫の形成には、脳と硬膜の間に隙間が生じる疾患、つまり 1)くも膜嚢胞(のう胞)や 2)特発性低髄液圧症候群などを合併している危険性があります。
飛行機頭痛 2012 2012/01/01
2012年に本HPで、飛行機頭痛について4回にわたり考えてみました。
(尚、2013年の国際頭痛分類第3版ベータ版により、飛行機頭痛が、二次性頭痛の【10.ホメオスターシスの障害による頭痛】の中に新しく分類され、診断基準も明記されたことをお知らせいたします。)
-起-
2010年10月にふらふら感を訴える60代男性の患者様がみえました。
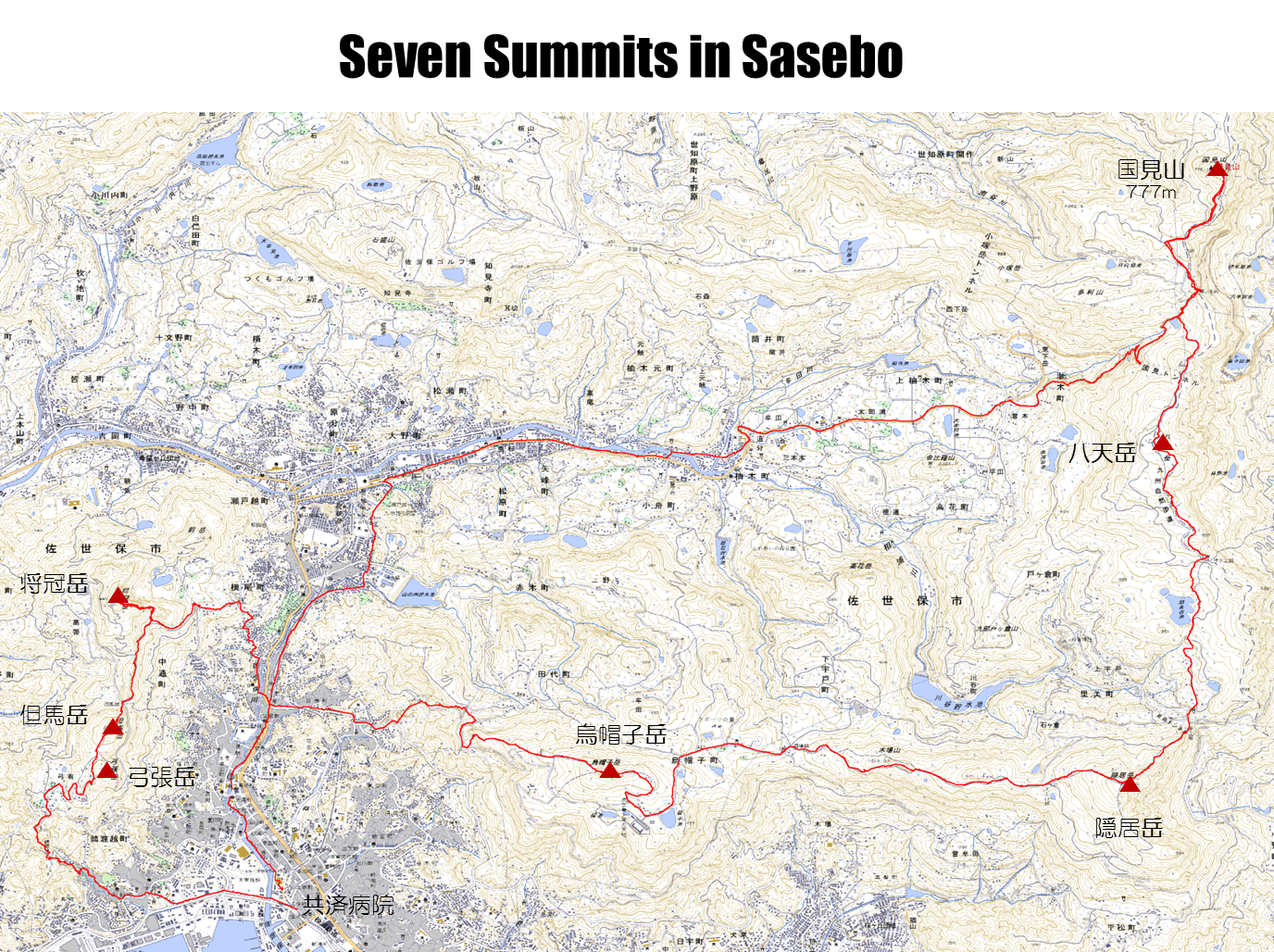
その方は山登り仲間と朝4時に出発し、愛宕山、赤崎山、弓張岳―将冠岳、烏帽子岳―隠居岳―八天岳―国見山を縦走、温泉に入って19時頃ビールを飲んでふらふらするという訴えでした。佐世保市街の周囲の山を全て周回するコースです。
あまりの距離に唖然としました、恐らく50㎞は超えています。60代で、まず、そのような無謀な?登山計画はやめるようにお話ししました。せめてその半分の距離にしたらとお勧めしました。
私は、弓張山系縦走 コース(鵜渡越―弓張岳―但馬岳―将冠岳)や裏烏帽子山系縦走コース(烏帽子岳―隠居岳―八天岳―国見山)は日頃歩いているので、その二つのコースを併せた佐世保一周コースには以前からとても興味がありました。
2011年12月になんとなく思いつき、それから準備に入りました。まず、1日で、共済病院→弓張岳→但馬岳→将冠岳→烏帽子岳→隠居岳→八天岳→国見山→共済病院、全コース徒歩とし、7つの山頂を超えるseven summits courseと名付けました。最後の国見山は、777mです。
縁起がよさそうなので2012年1月7日に登りました。予定は12時間です。途中、歩きながら考えてみるテーマは飛行機頭痛についてです、今年は4回に分けて飛行機頭痛について考えていくつもりです。
午前5時半出発、西九州の朝は真っ暗でヘッドライトをつけての行動でした。前半戦は暗闇の中の行動となり、周り(猪などの動物)に注意して飛行機頭痛など考える余裕はありませんでした。
弓張山系縦走コースを終え、俵町に降り、山手町、烏帽子岳を経て、日差しもでて段々暖かくなりました。
隠居岳からの展望はとても素晴らしく、目の前には、有田の街並みをはさみ青螺山の山並みが広がっています。隠居岳は縦走路の中核をなすいい山ですが、頂上は荒れ果て残念に思いました。隠居岳から北に延びる八天岳・国見山へのコースは穏やかな山道で、やっと飛行機頭痛について考えることができました。
飛行機頭痛はICDHⅡには分類されていないのではないでしょうか。しかし、airplane headacheという論文を時折見かけます。飛行機頭痛には、①日頃、頭痛のない人が、飛行機に乗った時だけ出現するもの(pure airplane headache)、②日頃から頭痛に悩まされている人が、飛行機に乗ると頭痛が出現するものに分けられます。今年考えるテーマは、①日頃、頭痛のない人が、飛行機に乗った時だけ出現するもの(pure airplane headache)についてです。
頭痛外来を行うようになってから、ずっと気になっていた私の飛行機頭痛について思い出すことにしました。それは、20数年前、私が医師になって3年目の頃のことです。東京の学会の帰り、羽田空港から長崎空港に向かう途中に経験しました。降下を始めた頃だと思います。急に前額部から目の奥が、ギューッと締め付けられるような激しい痛みでした。耐え切れない痛みで、死にそうな感じがしたのを今でも覚えています。雨の夜、真黒な大村湾にこのまま突っ込むのではないかと思うほどでした(この印象が強烈で降下時と覚えています)。
周りの人をみてみましたが、誰も痛そうではありません。私だけのようです。着陸後は、うそのように痛みはなくなっていました。もう一度経験しましたが、その後には経験していません。二回目の時も羽田から長崎行で降下時でしたが、一度目ほどは酷くなく、またかという感じでした。その頃、私なりに考えたことは、飛行機に乗る前から風邪気味で鼻水が出ていたので、副鼻腔の自然孔などを閉鎖して圧が変わる時に頭痛が生じたのだろうと考えていました。
圧が影響しているのだろう?ということで、今回の山行では、腕時計(プロテレック)で気圧を測りながら歩きました。標高とともに気圧は下がりますが、今回の山行の山頂での気圧を下に示します。この山行の最高地点である国見山(777m)では、933hPaでした。標高の高い2000m級の山では、気圧はもっと下がっています。
最近流行の山ガールの皆さん、片頭痛をおもちでしたら、気圧の変化に敏感で、きっと大変なのではないでしょうか。
国見岳登山口からの下山道は舗装されていてあまり楽しくはありませんでしたが、潜木町・柚木町を経て病院に着いたのは16時52分でした。
次回は、2011年の年末に敢行した飛行機の中の気圧測定結果などについて考えたいと思います。
| 共済病院 | 弓張岳 | 但馬岳 | 将冠岳 | 烏帽子岳 | 隠居岳 | 八天岳 | 国見山 | 共済病院 | |
| 標高(m) | 12 | 364 | 385 | 445 | 568 | 670 | 707 | 777 | 12 |
| 気圧(hPa) | 1023 | 983 | 980 | 972 | 960 | 948 | 941 | 933 | 1028 |
| 時刻 | 5:30 | 6:34 | 6:44 | 7:22 | 9:30 | 11:05 | 12:24 | 13:15 | 16:52 |
2012年1月7日天候晴れ・歩行時間11時間22分
2012年1月7日 GPSトラッキングデータより
-承-
前回、私が経験した飛行機頭痛を紹介し、佐世保市内の低山の山頂の気圧について調べました。私も経験したことがあり飛行機頭痛に興味をもち、自分のできる範囲の実験や観察を行ってきました。今回は、それらについて触れてみます。
航空機は、通常9000~12000メートルの高さを飛行します。上空は気圧が低いため機内は気圧調整され、0.7 ~ 0.8気圧程度になっているそうです。標高約2000~2500メートルの山に登っているのと同じ状態とされています。したがって離陸時の15~30分間に0.2 ~ 0.3気圧が下降し、着陸時の15~30分間に0.2 ~ 0.3気圧が上昇するという環境の変化になります。
気圧が低くなるとどんなことが起きるでしょうか。高い山に登るとポテトチップスなどの密封された袋がパンパンに膨れる事を見たことがあるかもしれません。飛行機の中でも同じことがみられます。
下にポテトチップスの袋の写真を呈示します。離陸前、ポテトチップスの袋に変化はありません。上昇し気圧が下がると袋はパンパンに膨れています。同じようなことが、ペットボトルでも経験されます。つまり、閉鎖腔は気圧が下がると膨らみます。逆に気圧が上がると潰れます。そして人間の体にも同じことが起きています。
 |  |
| 離陸前 | 上空で |
では、実際の飛行機の中はどうなっているでしょうか。2011年の年末に観察・気圧の測定をしてみました。長崎-羽田間で実行しました。気圧は腕時計(CASIO PRO TREK)で測定しました。
離陸前が1023hPaで、11時過ぎには751hPaとわずか30分くらいで272hPaも下がっています。約15分間751hPaと定常状態が続き、その後は降下に伴い約30分程度で気圧は上昇し着陸時には1018hPaに回復しています。
| 時間 | 10:16 | 10:52 | 10:53 | 10:54 | 10:57 | 11:00 | 11:04 | 11:20 |
| 気圧(hPa) | 1023 | 970 | 940 | 900 | 850 | 800 | 751 | 752 |
| 備考 | 離陸 | ベルトサイン 消える | 機長挨拶 | |||||
| 時間 | 11:35 | 11:37 | 11:40 | 11:42 | 11:45 | 11:47 | 11:49 | 11:57 |
| 気圧(hPa) | 762 | 800 | 850 | 884 | 927 | 940 | 1000 | 1018 |
| 備考 | 富士山が 見えます とアナウンス | ベルトサイン 点灯 A | B | 着陸 |
機内で気づいたことを記します。
ケース1:窓側に座っていた若い女性:彼氏にでも会いに行くのでしょうか。楽しそうな感じ、手鏡でお化粧をなおしていました。上空の定常状態(751hPa)から15分後の11時35分には762hPaと機内の気圧は上昇、11時40分気圧850hPaと急上昇し、可哀そうに髪を両手で掴みとても痛そうに下を向いています。先ほどまでのきれいな髪はぼさぼさです。私見ですが、気圧の変化に伴い頭痛を惹起したのではないでしょうか。私の気のせいかもしれません。
ケース2:11時45分927hPa(A時点)、私の前と後ろで子供が痛いと言って泣き始めました。子供さんは降下し始めに泣き始めることが多いようです。11時47分(B時点)、乗務員が急いでジュースを持って来て子供に飲ませています。大人は欠伸をしたり、唾をのみこんだり、耳抜きをしたり、耳管を開放することができます。子供は難しいのでジュースを飲ませて、飲み込む動作で耳管を開放させているのだと思います。11時51分には泣き止み、二人とも痛いとも言っていません。
私は、無意識のうちに嚥下運動や耳抜きを何度も繰り返していました。
天候が悪くなると気圧の変動がみられますが、飛行機の中ではこんなに急激に気圧が変化しています。
次回は、飛行機頭痛の論文をいくつか紹介したいと思います。
-追記-
最近、東京までジェット機で行ったときのデータや、プロペラ機で屋久島に行った時の
データを追加します。
1) 長崎空港→羽田空港
今回は、圧の変化に加えて、唾をのみこんだ回数、耳(鼓膜?)がガサッと音がして耳管が(自然に)
開放した回数、耳抜きの回数などを数えてみました。
離陸前(8時5分)は1011hPaで、8時27分752hPaまで下降し平衡状態に達しました。この間、
唾をのみこんだ回数は8回(22分間、0.36回/分)でした。鼓膜がガサッと音がした回数は、
左が8回、右が6回でした。
その後、752hPaの平衡状態は9時8分まで持続しました。この間は、唾をのみこんだ回数は
12回(41分間、0.29回/分)でした。鼓膜のガサッとする音は一度もありませんでした。
9時8分以降、機内の気圧は752hPaから急速に上昇しました。9時9分、耳がツーンと押された
感じがし、唾をのみこみました。左でガサッと音がして解放したことが確認されました。
降下時は唾の飲み込みは20回(27分間、0.74回/分)で、上昇時の2.1倍でした。
耳抜きは、降下時のみ2回行いました。降下時は、耳がガサッという音は、自然になったもの・
唾を飲み込んでなったものがあり、またその音の大小がありあまり正確には回数が把握
できませんでしたが、左は6回でした。それ以外に音以外で開いた感じがありました。
右では、音は5回で、回数は少なかったのですが音は大きかったようです。
降下時に、唾を飲み込む回数は1分間当たり倍以上となり、そして耳抜き行動を行なっていました。
降下時の圧変化は、私により負担になっているようでした。
| 時間 | 8:05-8-27(22分) | 8:27-9:08(41分) | 9:08-9:35(27分) |
| 機内の気圧(hPa) の変化 | 1011hPaから 752hPaまで下降 | 752hPaで 平衡状態 | 752hPaから 1013hPaまで上昇 |
| 唾をのみこんだ回数 | 8回(0.36回/分) | 12回(0.29回/分) | 20回(0.74回/分) |
| 右耳でガサッという音の回数 | 6回 | 0回 | 5回 |
| 右耳でガサッという音の回数 | 8回 | 0回 | 6回 |
| 耳抜き回数 | 0回 | 0回 | 2回 |
2) 福岡空港→屋久島空港
地上では、1010hPaでしたが、上空でも957hPaまでしか下降しませんでした。帰りも同様でした。
機長からの報告では4600mの高度を飛行しているというアナウンスでした。
プロペラ機ではジェット機よりも低い高度を飛行し、圧の変化も小さいようです。
-転-
airplane headacheは、国際頭痛分類第2版には分類されていません。今回は、これまで報告
されている飛行機頭痛に着目します。私は、airplane headacheには、
①普段、頭痛のない人が飛行機に乗った時だけ頭痛が生じる場合と、
②日頃、片頭痛や緊張型頭痛、群発頭痛を持っている人が飛行機に乗ると頭痛が生じる場合が
あると考えてきました。
類似の考えの論文もありますが、全く一緒というわけにはいかないようです。
参考文献を下に示します。
| 著者 | タイトル | |
| 1 | Evans RW | Airplane descent headaches. Headache 2007;47:719-723 |
| 2 | Berilgen MS | Headache associated with airplane travel: report of six cases. Cephalalgia 2006; 26: 707-711 |
| 3 | Atkinson V | An unusual case of an airplane headache. Headache 2004; 44: 438-439 |
この中では、1の論文が私の考えに近いと感じました。最初にこの論文を紹介します。
この論文では4人の患者を紹介しています。全例ともjet airplaneで降下時の激しい頭痛です。
症例1:
22歳男性、non-smoker、前兆のない片頭痛が10年来あります。
ジェット機で、降下時に、突然、左こめかみを中心とした痛みが出現し、
shooting pain (刺痛・電撃痛)、bursting pain(爆発するような痛み)と表現しています。
この痛みの程度は10/10で、耐えられないもので約25分間続いています。
光過敏や音過敏、吐き気などはありません。
→つまり、片頭痛の既往がある人に、片頭痛とは異なる頭痛が生じたことを示しています。
症例2:
23歳女性、non-smoker、頭痛の既往はありません。
降下時に、突然、stabbing pain(刺すような痛み)が頭頂部を中心に出現し、
15分間続いています。その他の症状はありません。
症例3:
32歳の女性。Non-smoker。
飛行機に乗った最初の頃、降下時に右頭頂部に締め付けられるような頭痛を感じています。
これは4時間から12時間継続し、頭痛の程度は2-5/10でそれほどひどくはなく、
音過敏があり、時折、目がかすむようなこともあったそうです。
そのようなことが続き、現在は、毎日7-10/10程度の頭痛があり、
さらに飛行機に乗ると降下時に10/10の頭痛となっています。
効果がなくてもイブプロフェン200㎎を1日に12回内服するということです。
→飛行機に乗るようになって片頭痛が生じるようになり、それが慢性片頭痛・薬物乱用頭痛となり、
飛行機の降下時には頭痛がひどくなるという病態です。
症例4:
26歳男性、non-smoker
降下時に、突然、突き刺すような頭痛が左眼窩上部に出現しています。
10/10の痛み、拍動性ではなく、流涙、吐気、めまいなどはないそうです。
着陸後、約30分程度で消失しています。私の経験した頭痛は症例4に類似しています。
これらについて、著者はいくつかの意見を述べています。そのうちのいくつかを取り上げます。
①片頭痛は、飛行機旅行で惹起されることがあります。
筆者らの施設で385人の片頭痛患者を調べたところ、8%で飛行機旅行中に片頭痛の出現を
認めています。これは、飛行機による影響も考えられるし、旅行という特殊な状況
(睡眠不足とからストレス)などの影響も考えられると述べています。
→片頭痛を持った人は、飛行機に乗ると片頭痛が生じやすいことを示しています。
②飛行機頭痛が生じる機序を筆者らは次のように考えています。気圧の変化による副鼻腔の
異常が頭痛を惹起するという考えです。降下時に、機内で急激な気圧上昇がおこり、このために
sinonasal barotrauma(気圧の変化による副鼻腔損傷)が生じると考えられています。
つまり、閉塞された副鼻腔内の圧は、比較的低く保たれ、降下時に機内の気圧の相対的な上昇に
伴い、内向きにvacuum effect(“the squeeze”)が生じ、このために、副鼻腔内の組織に
影響を与え、痛みが出現するという考えです。
40年くらい前の私が小学生の頃の話ですが、湯呑み茶碗を口の周りにつけて、落ちないように
湯呑みの中の空気を吸い込むという遊びをやっていました。湯呑みが落ちないで、どれくらい
いられるかというたわいもない遊びです。それはそれで面白かったのでしょうが、その後の結果は
悲惨なものでした。
口の周りが赤紫になって、石鹸で洗っても落ちるはずもなく、恥ずかしくて学校にも
行きたくありませんでした。つまり、陰圧で口の周りの皮膚を吸い続けたようなもので、
赤いのは静脈性浮腫?、口の周りが赤くなっていたのです。
こういうことが降下時に副鼻腔に起きている(sinonasal barotrauma)ということです。
通常は、副鼻腔と外気は交通していて、こういうことが起きないように、ある動き
(唾をのみこむ、欠伸をする、耳抜き)をしています。
この②の考えは、私の考えとほぼ同じです。著者の考えと私の考えの違いは、
本質は同じだと思いますが、表現が違うところです。筆者らの考え方は副鼻腔内圧が低く、
そのために吸い込むという考え方です。
それはそれでいいと思いますが、なかなか痛そうに感じません。副鼻腔の周りの圧が
相対的に高くなり、そこを外から内向きに押すと考えると、神経が押されてとても痛そうに
感じます、どうでしょうか。着陸し、圧の平衡状態が回復すると、痛みが消失すると考えられます。
2.の論文では、6例の飛行機頭痛を紹介しています。
日頃は頭痛のない人が、離陸時あるいは着陸時に15-20分の激しい頭痛を経験するというものです。
機序としてやはりbarotraumaにより、篩骨神経が影響されるという考えです。
3.の論文は1例報告です。28歳の男性で、上昇時と降下時に激しい痛みを訴えています。
上昇時に、前頭部と眼につきさるような突き刺すような激しい痛みが始まり、
これは上空では一旦落着き、降下時にもう一度、頭痛が出現しています。光刺激・音刺激は
ありません。2や3の論文は、降下時に加えて昇行時にも痛みが出現しています。
昇行時にも生じているところが、1の論文と異なる点です。
-結-
最後に飛行機頭痛についての私なりのまとめを呈示します。
(概念)
飛行機に乗った際に生じる頭痛で、大きく次の二つのタイプがあります。
①これまでに頭痛の既往がなく、飛行機に乗った時、特に降下時に生じる頭痛。
1)激しい頭痛、2)部位は前頭部、眼窩、側頭部に多い
②片頭痛や群発頭痛の既往があり、飛行機に乗った時にこれらの頭痛が惹起される。
純粋な飛行機頭痛は①に相当します。こちらは国際頭痛分類の二次性頭痛に分類すべきものではないでしょうか。
(純粋な飛行機頭痛の起こる機序)
離陸前は日本の平地では1010hPa程度です。上昇時に機内の気圧が下がり、上空では機内の気圧は
低い状態(800atm前後)で平衡となります。降下時には、800hPaから1000hPaまで急速に上昇します。
低い気圧で平衡状態となった閉鎖腔の副鼻腔の外の気圧が相対的に高くなり、そこを外から内向きに
押すと考えると、神経が押されて痛くなるのではないでしょうか。着陸し、圧の平衡状態が回復すると、
痛みが消失すると考えられます。論文ではbarotraumaという単語が頻用されています。
(純粋な飛行機頭痛の治療&予防)
副鼻腔炎や中耳炎を患っている場合は、特に上記の状態が起こりやすいと考えられ、そういった疾患は
前もって治療しておく。風邪などで鼻炎、鼻つまりも同様と考えられます。
嚥下運動や耳抜きなどで副鼻腔や中耳内の圧を機内の圧と一緒に保てるようにする。
具体的には、唾を飲み込む、アメをなめる、欠伸をする、このような所作により耳管が開放されます。
耳抜きはいい方法ですが、あまり強くやりすぎるのはいけません。
かなりの確率で、激痛発作が来ることが予想される場合には、ロルフェナミンなど鎮痛剤を
離陸する前に内服し上空で薬剤の有効濃度がすでに到達するように工夫し、さらに痛みが来た場合には
追加することなどが有効ではないかと考えています。
未だ不明な点が多く、純粋な飛行機頭痛の治療&予防が研究・開発されることが待たれます。
(片頭痛や群発頭痛のある人で、機内でこれらの頭痛が惹起される場合の治療)
もちろん、片頭痛や群発頭痛が機内で惹起される方は、片頭痛発作時の薬剤あるいは群発頭痛
発作時の薬剤の準備が必要です。
付録
①飛行機頭痛で、私が気圧にこだわったわけがあります。医師になって3年目の頃のことです。
巨大動脈瘤の治療を施行した患者様がおられました。現在のような血管内治療もなく、
当時は頚部で内頚動脈を螺子で数日かけて閉塞させていく治療でした。最後の一螺子をしめると
失語症と右片麻痺が出現し、緊急血栓除去術を行いましたが、その時には栓子が末梢に
移動していました。麻痺は改善しましたが、軽い失語症が残存しました。
そこで高気圧酸素療法を
施行することになりました。今の高気圧療法の器械はカプセルのような形ですが、その当時の
器械は潜水艦のような形で、医師が一緒に入る規則になっていました。
私が一緒に入ることになったのですが、気圧を上昇していくと、右耳付近が激しく痛くなり白旗を
上げ出してもらいました。私の考えでは、右の耳管がおそらく狭窄していて、気圧上昇に伴い、
鼓膜に圧が加わり中耳を刺激し激しい痛みが出現したのです。耳鼻咽喉科では、この状態を
【気圧上昇に伴う中耳炎】と診断されています。
私は、患者様と一緒に入らないといけませんから、
耳鼻咽喉科の先生に相談して右の鼓膜に小さな穴をあけてもらい、気圧が上昇しても中耳内の圧も
鼓膜の外の圧と同じになるようにし、無事治療を続けることができました。その患者様の症状は、
徐々にではありましたが、軽快しました。
鼓膜に小さな穴をあけるときは、ゴソッととんでもない大きな音がしました。この程度の小さな穴は
自然に塞がり何の問題もありませんでした。この時の高気圧治療の器械に入った時の頭痛
(耳付近の痛み)と私の飛行機頭痛が頭の中ですぐに結びついたのです。
②今年の夏、一人の患者様が、海外に旅行に行って頭が痛くなり頭痛外来を受診されました。
目的地までは飛行機で移動し、飛行機の降下時に左耳付近、左耳の後ろあたりに激しい痛みが
生じたそうです。着陸後は、改善はしたものの頭痛が続き、帰ってきてからも同じような状態が
つづくというお話でした。
もともと、中耳炎があったそうです。画像診断も行いましたが、頭蓋内に異常はありませんでした。
もうお気づきでしょうが、この患者様は、中耳炎などにより、耳管が狭窄・閉塞しており、
気圧の変動の関係で、中耳などに負担がかかり、痛みが出現したものと考えられます。
つまり耳の痛みを耳の後ろ付近が痛いという訴えで頭痛外来を受診されたのです。この疾患は、
耳鼻咽喉科領域では【航空性中耳炎】という診断名がついています。同じような理屈で
涙腺、歯などの痛みも飛行機に乗って生じることがあります。
薬物乱用頭痛の論文の紹介2013/11/01
頭痛で苦しんでいる患者様が大勢いらっしゃいます。
診断治療が困難な疾患に慢性連日性頭痛があります。
慢性連日性頭痛には、慢性片頭痛、薬物乱用頭痛、慢性緊張型頭痛などがあります。
薬物の使用が過度な場合、薬物乱用頭痛が疑われます。
薬物乱用頭痛には、大きく2つに分けられると考えられています。
1)病態を説明し、薬物の乱用が中止できるもの
2)薬物乱用が中止できないもの
実際には、この中間の患者様もいらっしゃいます。
患者様から、頭痛はどうしてなるのですか、とか、頭痛は治りますか、などという質問を受けることがあります。このような場合、一般的な説明を行います。実際にはあまりよくわかっていません。患者様は苦しんでいます。そして、脳は苦しんでいます。
薬物乱用頭痛で苦しんでいる脳の世界を、薬物乱用頭痛の論文から覗いてみます。
薬物乱用頭痛に関する一つの論文を紹介します。2005年に発表され、日本で薬物乱用頭痛に関する発表やテキストに必ずと言っていいほど、今なお引用されています。脳神経外科の手術ばかりやってきた私には、最初の2年くらいは、この論文は読もうとしても、全く歯が立たずそのまま放置してありました。その後、何回かこの論文を読んでみようと試みましたが、やはりわかりませんでした。最近、なんとなくですが、わかってきたような気がします。
はじめに
薬物乱用頭痛は、臨床上重要な疾患です。片頭痛や緊張型頭痛の患者様が鎮痛薬などを定期的に使用することによりその頭痛を悪化させるものと理解されています。この論文では薬物乱用頭痛のメカニズムについて考察しています。
薬物乱用頭痛を引き起こす薬剤について
薬物乱用頭痛は、複合鎮痛剤、バルビツレート、オピオイド、エルゴタミン製剤、アスピリン、NSAIDs、カフェイン、トリプタンの乱用によって出現します。薬物乱用頭痛が出現するまでの期間は、トリプタンで1.7年、エルゴタミン製剤で2.7年、鎮痛剤で4.8年と報告されています。
片頭痛を有する患者が、オピオイドを毎日使用した場合、それがどのような使用目的であっても、慢性連日性頭痛のような変容性片頭痛になる危険性があると報告されています。つまり、片頭痛を有する人がオピオイドを腰や、肩や膝など他の部位の鎮痛に使用しても薬物乱用頭痛になる危険性を秘めています。
薬物乱用頭痛における感作について
感作とは、同じような刺激が繰り返し行われることにより、その刺激に対してより強く反応することです。
薬物乱用頭痛の特徴は、頭痛の頻度が増すこと、頭痛の範囲が広がること、皮膚アロディニア(通常では痛みを感じないような刺激に対して痛みを感じる事)などです。これらは、中枢性感作により生じていると考えられています。中枢性感作は三叉神経系から中脳水道灰白質にまで拡がっていきます。もっと拡がっていると考えられます。
三叉神経への度重なる刺激は、三叉神経尾側核の神経に機能的変化をきたし、侵害刺激に対する閾値を下げ、受容野の拡大をきたします。この結果、痛みの頻度が増し、痛みの範囲も変化をきたすと考えられます。一方、内因性疼痛コントロールシステム(後日記載)の抑制は、中枢性感作を容易にします。
しかし、片頭痛の頻度が増すこと、薬物使用頻度が増すこと、いずれの結果で感作が生じるのかは明確ではありません。
トリプタンを慢性的に使用すると、脳表や脳幹のセロトニン受容体に変化をきたすことが知られています。セロトニンによる疼痛コントロールシステムの可塑性は、容易に感作を惹起し、慢性頭痛を引き起こします。セロトニンは片頭痛のメカニズムに重要な役割を果たすばかりでなく、薬物乱用にも関与していると理解されています。
側坐核、背側線条体、前頭前野なども、感作に重要な役割を果たしています。この際の神経伝達物質はドーパミンと考えられています。ドーパミン、セロトニンの両者のバランスが、薬物乱用頭痛のおける感作に重要な役割を果たしていると考えられています。
片頭痛の病態及び片頭痛が薬物乱用頭痛に変容していくことについて、解剖学的構築の考察
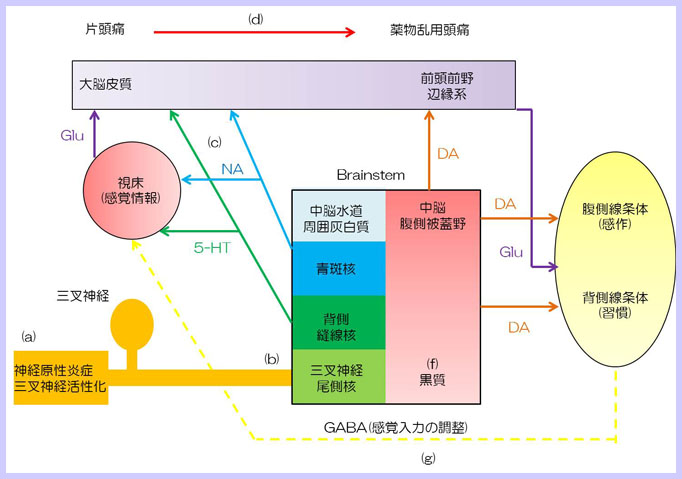
(a) 片頭痛は、神経原性炎症と三叉神経の活性化によって生じます。
(b) これらの刺激は、脳幹に拡がっていきます。すなわち、三叉神経尾側核(TNC)、背側縫線核(DRN)、青斑核(LC)、中脳水道周囲灰白質(PAG)に拡がっていきます。
(c) これらの核に伝わった刺激は、さらに視床や大脳皮質に拡がっていきます。
この際、背側縫線核(DRN)からの神経伝達物質はセロトニン(5-HT)で、青斑核(LC)からの神経伝達物質はノルアドレナリン(NA)です。
(d) 頭痛治療薬の過度の使用をともなった頻度の多い片頭痛発作は、次第に薬物乱用頭痛に変容していきます。
(e) この変容には中枢性感作が重要と考えられています。中枢性感作は、脳幹(中脳腹側被蓋野)から前頭前野と辺縁系、そして腹側線条体への刺激によるものと考えられます。この際の神経伝達物質はドーパミンです。つまり、中枢性感作は、ドーパミンのリリースが引き金となっていると考えられています。
(f) 中枢性感作に引き続き、薬剤の過度の使用という習慣が維持されるようになります。これには、中脳の黒質からの背側線条体へのドーパミンの作用が重要とされています。
(g) 線条体は、視床において他の感覚入力を調整しています。この際の神経伝達物質はGABAです。
わかりにくいので、一気に垣根を取り外してしまいます。片頭痛は、三叉神経血管系の神経原性炎症によって生じるとされています。そして頻回の片頭痛発作と薬物の乱用によって中枢性感作が生じやすいと考えられています。これには、PAG、DRN、LCなどの下行疼痛抑制系とVTAから前頭前野や側坐核への脳報酬系がからんでいるのではないでしょうかという考察です。
5年間、手元にあった論文にはこんな感じの内容でした。その他に心因的な共存症や生化学的機序も記載されていましたが省略いたします。
出典文献を示します。
| 著者 | タイトル | |
| 1 | Calabresi P | Medication-overuse headache: similarities with drug addiction. Trends in Pharmacol Sci 2005;26:62-68 |
慢性連日性頭痛~苦悩&苦脳~2014/03/01
頭痛外来には、慢性の頭痛に悩まされて受診される患者様がいらっしゃいます。
慢性連日性頭痛は、Silbersteinにより提唱されました。
この中で、頭痛外来で、よく受診されるのが薬物乱用頭痛と慢性片頭痛です。
患者様はこの頭痛に苦悩されています。そして患者様の脳も苦しんでいます。
第一に困るのは、その診断です。
国際頭痛分類第2版(ICHD-2)により診断してきました。2013年に国際頭痛分類第3版β(ICHD-3β)が公表されました。
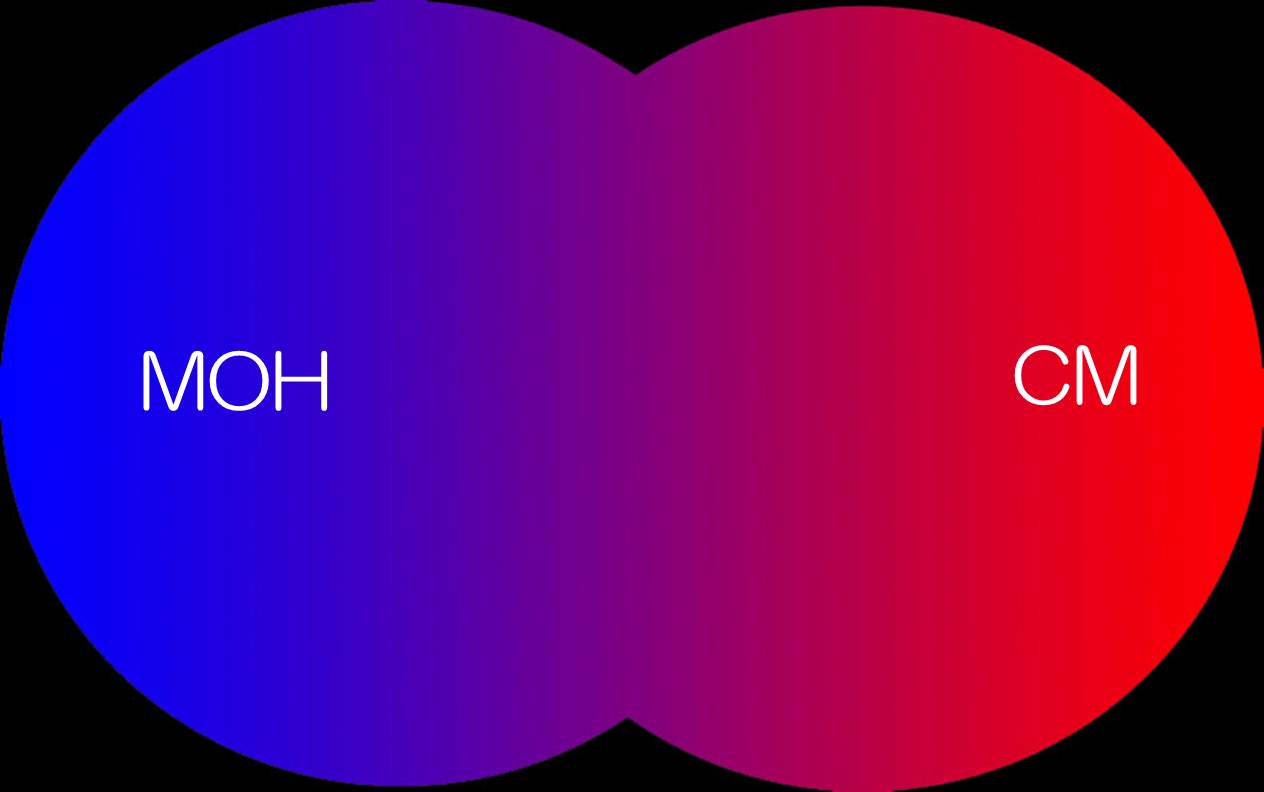
国際頭痛分類第2版(ICHD-2)の薬物乱用頭痛(MOH)と慢性片頭痛(CM)の診断基準を示します。
 |  |
診断基準は、かなり良く似ています。そもそも、MOHにおいて、薬剤を定期的に使用とは、どういう状態でしょうか。 そして、月に10日以上、トリプタンを使用し、しかも効果のある場合は、なんと診断すればいいのでしょうか?月に10日以上、トリプタンを使用するということは、慢性片頭痛の診断基準のD項目に相当しません。しかし、効果があるということは薬物乱用頭痛の診断基準のC項目には相当しないと思います。上記診断基準には抜け道がたくさん? 私は、なんとなくこんなイメージを持っていました。
 |
次に国際頭痛分類第3版β(ICHD-3β)を示します。
 |  |
今回の薬物乱用頭痛の診断基準から、頭痛は薬物乱用により悪化出現したという表現が削除されています。慢性片頭痛の診断基準から、薬物乱用が存在しないという表現が削除されています。
いよいよ、私にとって初診時に診断をすることは困難になりました。こんなイメージです。
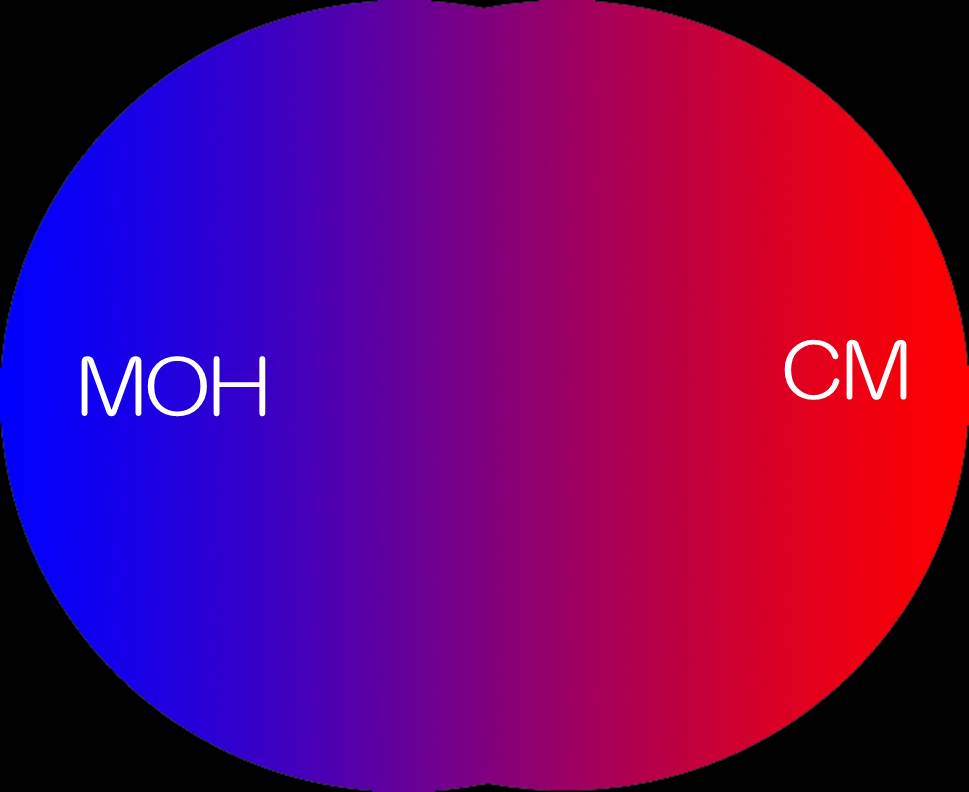 |
今回の国際頭痛分類第3版β(ICHD-3β)からβがとれ、公のものとなるのは2016年だそうです、それまでに見直しもあるそうです。
次に当院頭痛外来を2009年春に開設以降、2013年末まで5年間のこの2疾患の診療結果について示します。
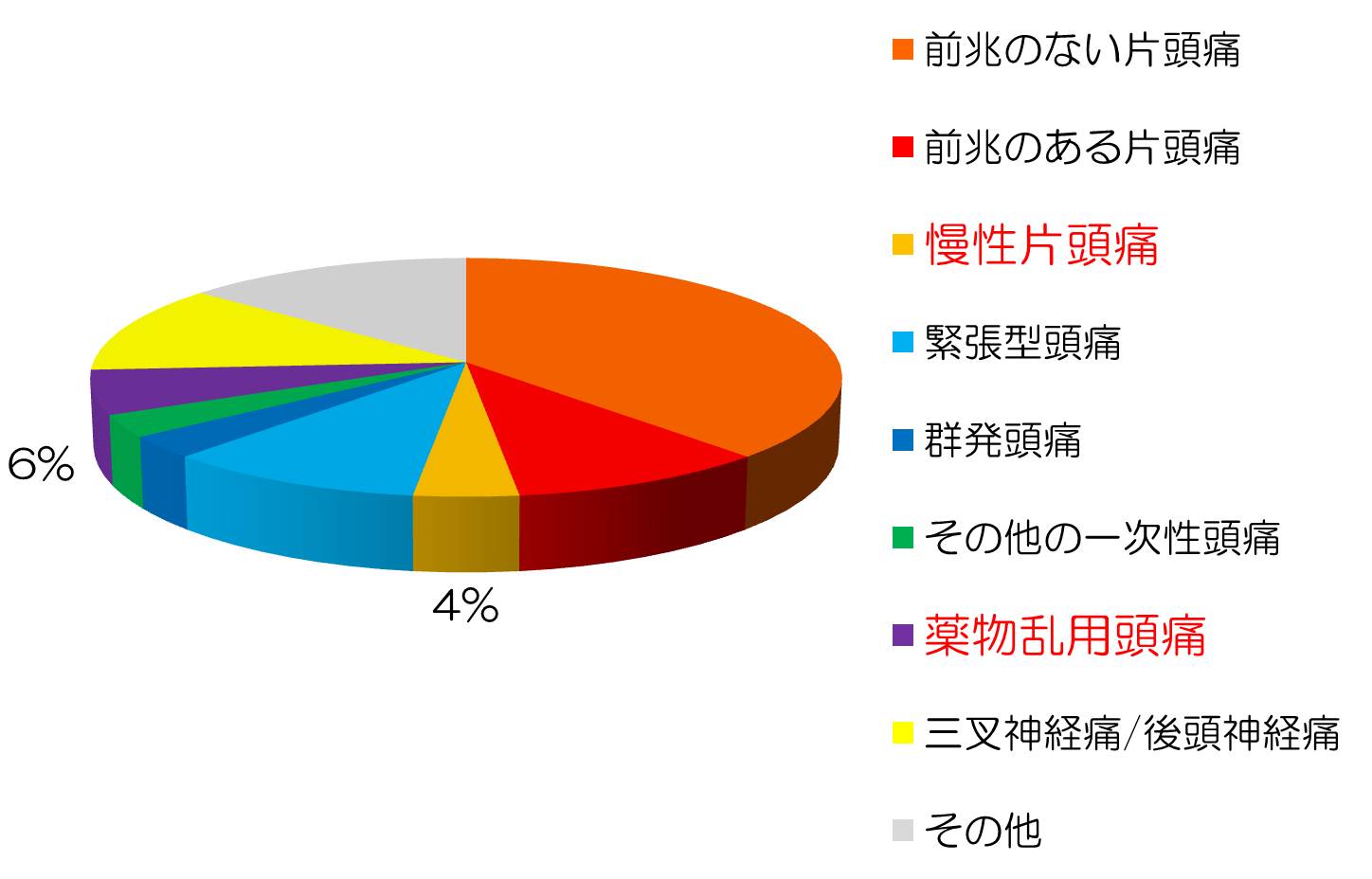
①ICHD-2を用いて初診時の第一診断による割合を示します。薬物乱用頭痛と診断したのが約6%で、慢性片頭痛と診断したのが約4%です。
 |
②性比を示します。圧倒的に女性に多くなっています。
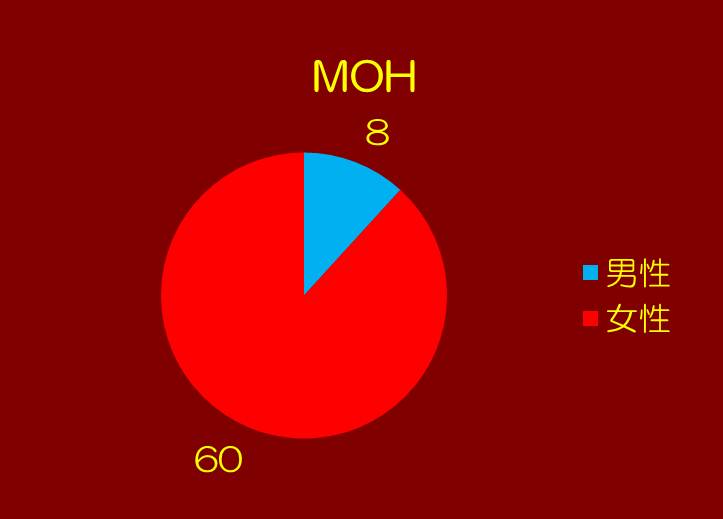 | 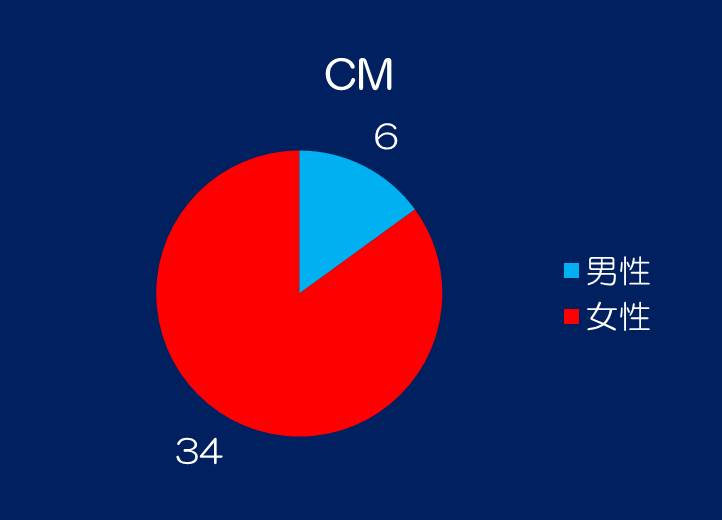 |
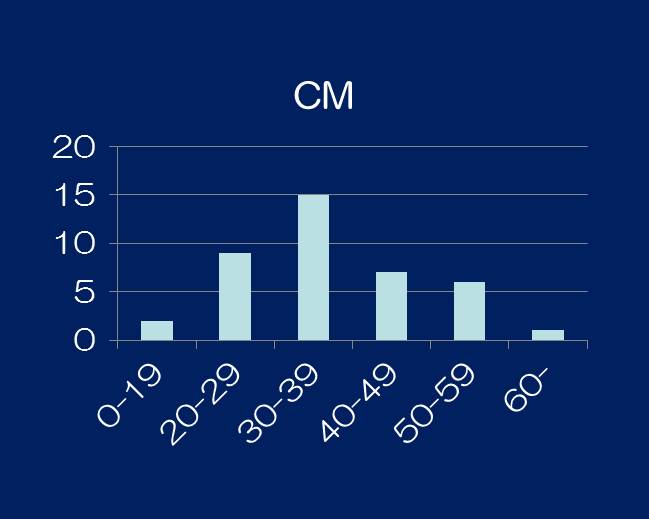
③年齢分布を示します。MOHでは、ピークが40歳代(平均年齢44歳)で、CMでは、ピークが30歳代(平均年齢38歳)となっています。
 |  |
④内服薬剤の入手方法を示します。両群ともに42%の患者様が市販薬を購入されています。
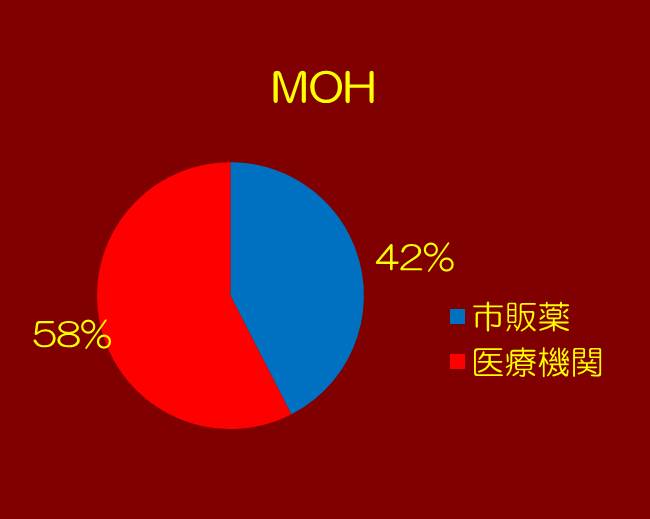 | 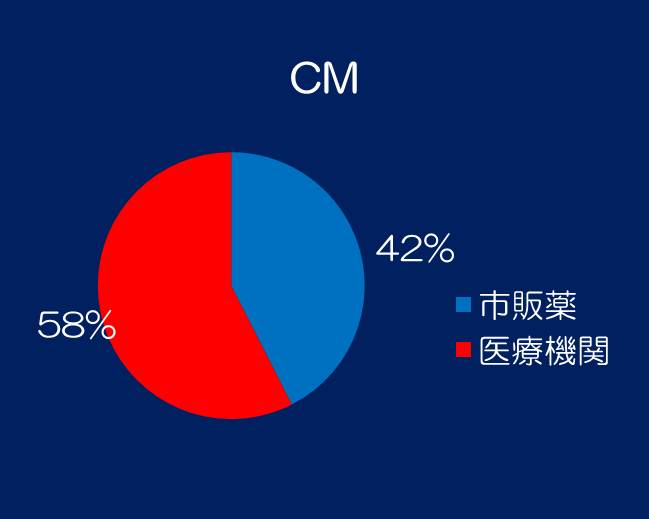 |
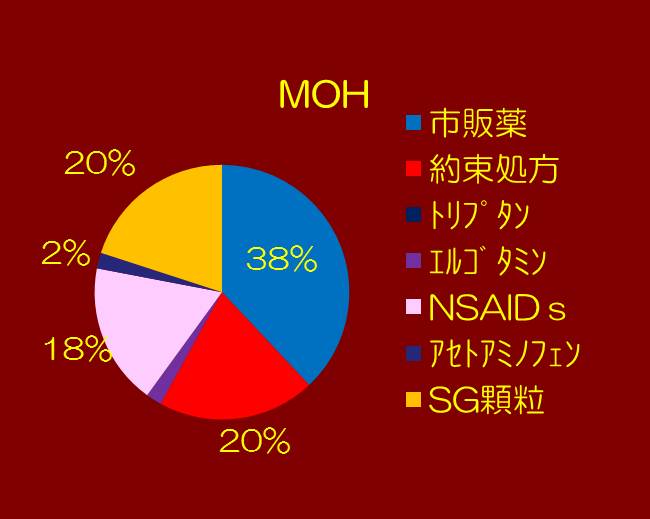
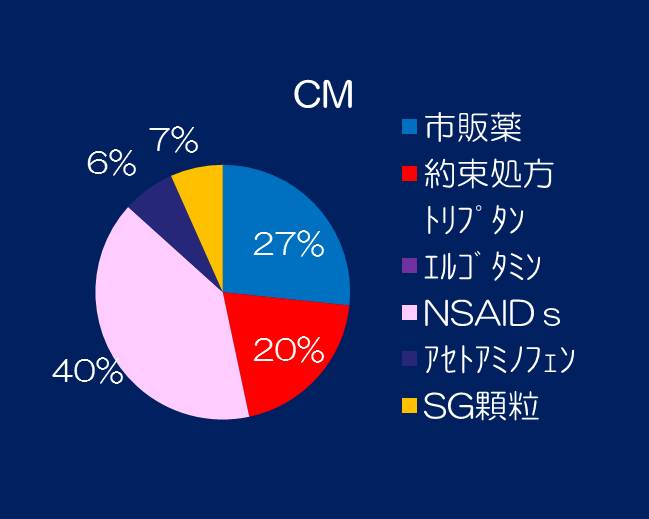
⑤医療機関から処方された鎮痛剤などの種類を詳しく示します。MOHでは、約束処方やSG顆粒など、複合鎮痛剤の処方が増加しています。それに対し、CMでは単一の鎮痛剤の割合が多いようです。
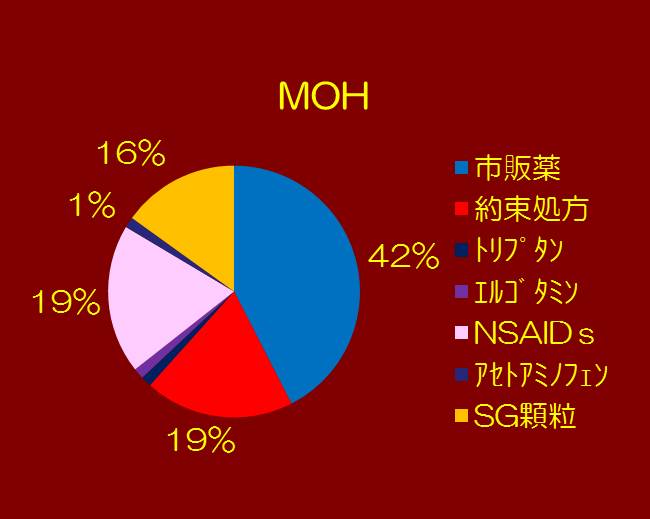 | 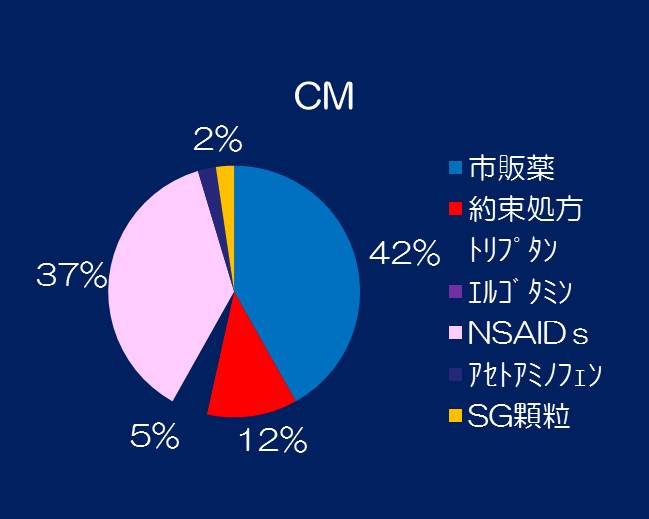 |
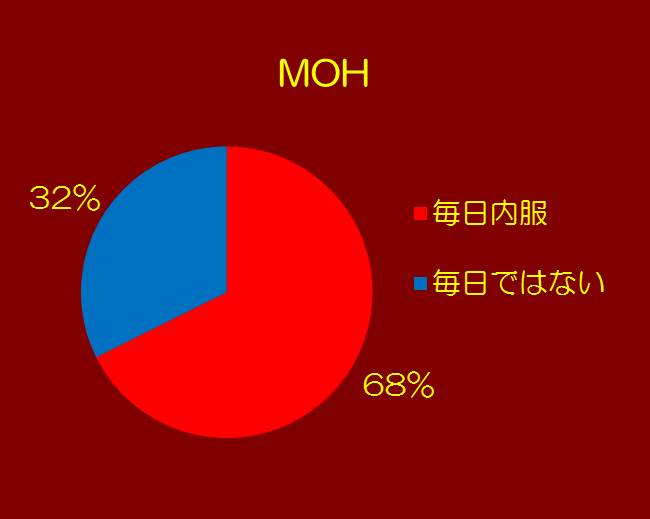
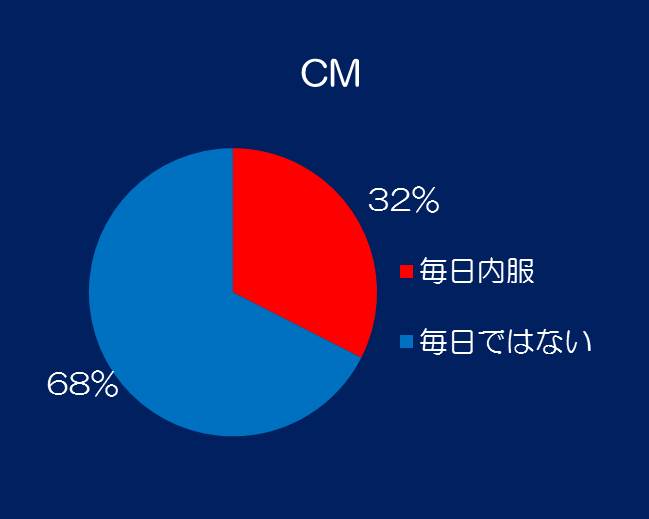
⑥薬剤を毎日内服する割合を示します。MOHでは薬剤を毎日内服するのは68%で、CMでは32%です。MOHでは毎日薬剤を内服する傾向が強いようです。
 |  |
ここからは、治療およびその結果を示します
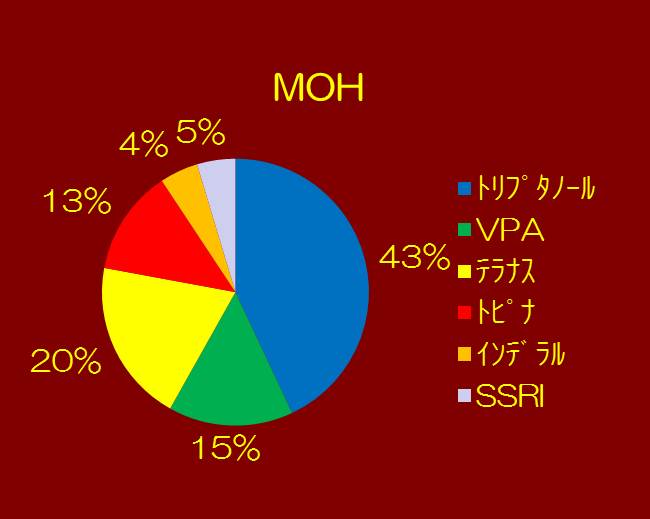
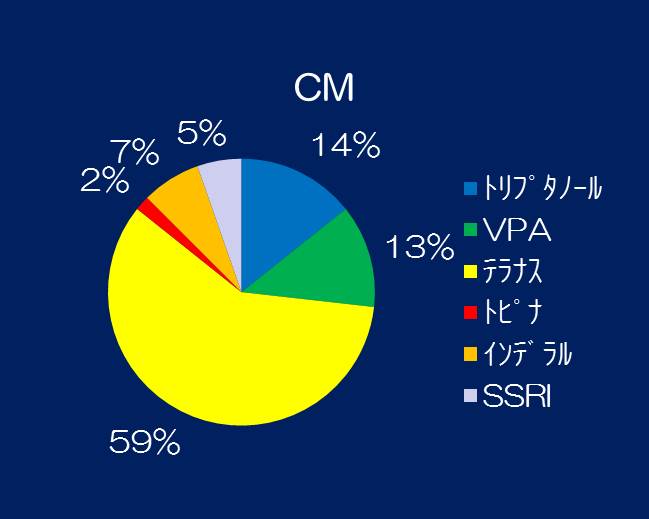
①当院では、どのような予防薬を使用したかを示します。MOHでは、トリプタノールが最も多く、CMでは、テラナスが最も多くなっています。
 |  |
②治療結果を示します。
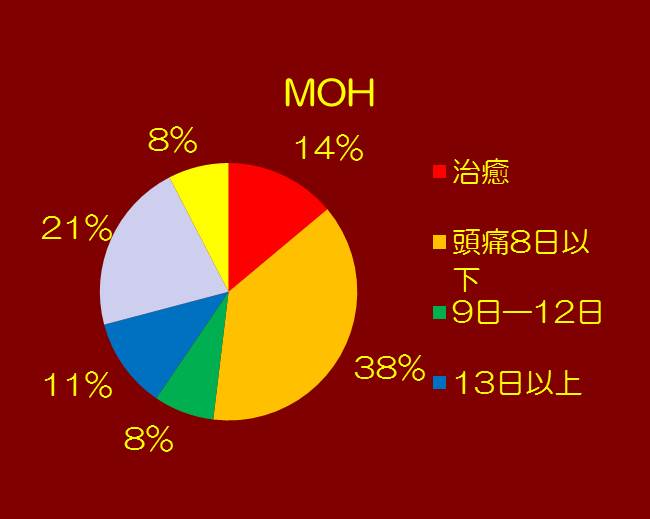
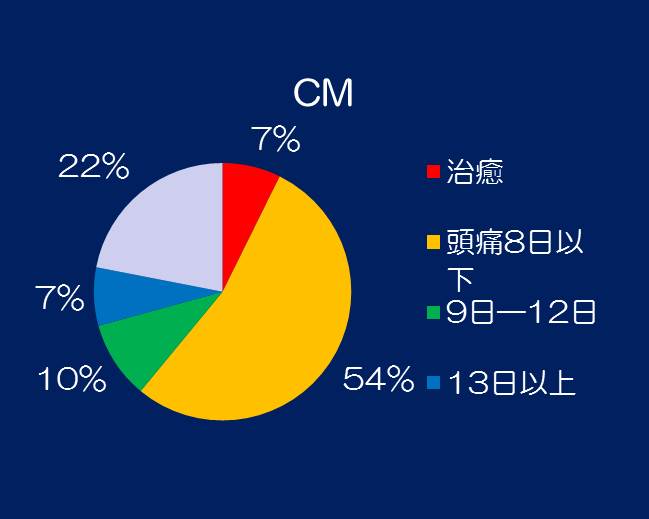
ⅰ)最初に、頭痛日数に着目します。治療以前は頭痛が15日以上あり、頭痛日数が8日以下となったものを改善とします。改善は、MOHでは52%、CMでは61%です。
 |  |
ⅱ)頭痛の程度まで加味すると、改善度は若干アップし、それぞれ60%、68%となります。
 |  |
ⅲ)その他
上記の図をみてみるとドロップアウトが22-21%です。
MOHでは説明のみに終わったものが、8%を占めています。これは、病態の理解が得られず、残念ながら、治療に至らなかったことを示しています。
【まとめ】
①MOHおよびCMとも女性に多く、MOHではCMよりも8歳位年齢が上がっています。これは、薬剤を飲み続け、効かなくなっていくのを示しているのではないでしょうか。
②内服している薬剤も、MOHでは複合鎮痛剤の使用が増えています。つまり、当初は単純鎮痛剤で対応していたものが、効果が少なくなり、複合鎮痛剤の使用が増えていったものと考えられます。
③頭痛外来で、治療効果があったものは、MOHで60%、CMで68%にすぎません。ドロップアウトの群にはよくなって来なくなった患者様がいらっしゃるので、若干数値はあがるのかもしれませんが、よくならない患者様も多くいらっしゃることを示しています。
慢性連日性頭痛にどうしてなるのでしょうか?
慢性連日性頭痛にならないようにするにはどうすればいいのでしょうか?
慢性連日性頭痛の治療はどうしたらよいのでしょうか?
慢性疼痛の論文の紹介~パラダイムシフト~2014/05/01
最近、二人の女性が発表された慢性疼痛に関する論文を読みました。
仙波恵美子先生の論文と半場道子先生の論文です。
慢性(連日性)頭痛の診療にも関係していて、大変参考になりました。
最初に仙波先生の報告を紹介し、次に半場先生の論文を紹介します。
仙波恵美子先生の報告
1)慢性痛による情動障害・睡眠障害と脳報酬系(ペインクリニック34:611-623, 2013)
慢性疼痛患者はうつや睡眠障害を伴うことが多い。
ストレスによっても痛みは増強し、うつが発症し、痛み、ストレス、うつが相互に関連すると述べられています。内容がかなり難解なので、私がなんとかわかる部分についてのみ触れたいと思います。
痛みの中枢回路
痛みの中枢回路には、外側系と内側系が知られています。各々が示しされています。
外側系は、脊髄後角Ⅴ層のWDRニューロンが視床後腹側核を経て体性感覚野に投射する経路で、痛みの識別に関与しています。
内側系は、視床の内側核群を経て前帯状回や島皮質に投射する経路で、痛みの情動や認知に関与しています。
さらに痛みの情報は、体性感覚野から後部頭頂葉、島皮質に投射し、島皮質から扁桃体に投射することが明らかになっていると述べてあります。
内側系や外側系の視床を介する経路はserial pathway(脊髄-視床路)として紹介されています。
一方、脊髄後角Ⅰ層のNSニューロンから、網様体や脚傍核、視床下部、扁桃体を経て島皮質や前帯状回に到達する経路も明らかになっており、pararell pathway(脊髄-脚傍核-扁桃体路、脊髄-脚傍核-視床下部路)として紹介されています。こちらの経路は視床を経由しません。
前帯状回からは前頭前野、扁桃体に投射し、前頭前野、扁桃体、視床下部からは側坐核に投射し、前帯状回の錐体細胞から視床下部、扁桃体、中脳中心灰白質にも投射します。前帯状回は、前頭前野などのmotor planningに関連し痛みや情動に伴う行動の選択にも関わっていると述べています。
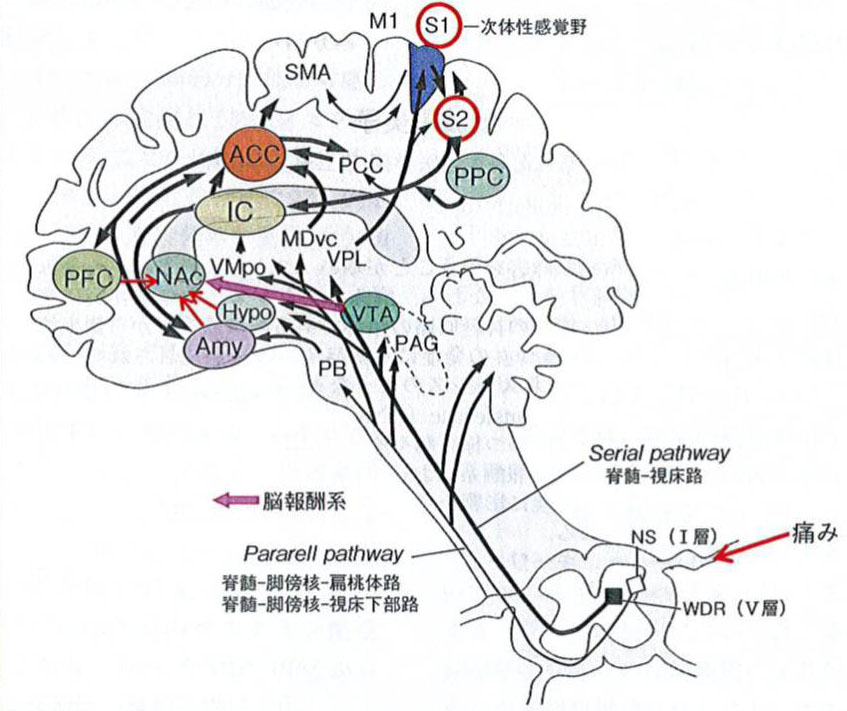 |
情動の神経回路
情動の発現には、腹側被蓋野から側坐核に至るドーパミン作動性の脳報酬系が重要な役割を果たすと述べています。側坐核には、前頭前野、海馬、扁桃体、視床下部、背側縫線核、青斑核などからの投射があります。
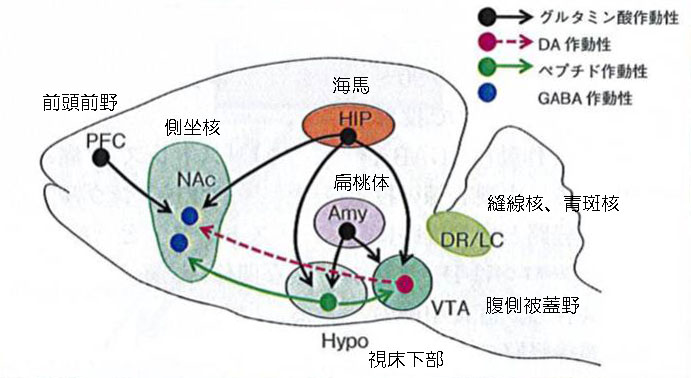 |
脳イメージの研究では、急性疼痛では、体性感覚野、前帯状回、島皮質の脳血流が増加するのに対し、神経障害性疼痛では、前帯状回、島皮質の脳血流が増加します。
⇒急性疼痛では、痛覚識別の領域までの道筋が必要ですが、神経障害性疼痛ではそういった道筋とは別に、痛みの情動や認知といった領域で痛みを感じるということでしょうか。
さらに、情動障害・睡眠障害と脳報酬系、ストレスについて述べてあります。脳報酬系が快情報ばかりでなく、不快情報に関与していることを示し、慢性疼痛やストレスなどの不快情報が、報酬系においてエピジェネティクス修飾による遺伝子発現の変化を惹起しうつや睡眠障害をきたしているのではないかと考えています。痛みを放置することによるエピジェネティクス修飾と情動障害・睡眠障害の関係を明確にすることが今後必要だと述べています。
2) 痛みが慢性化する脳メカニズム(神経内科78:348-360, 2013)
持続する身体の痛みが、前帯状回や扁桃体などでグルタメート作動性神経伝達を強め、GABA作動性伝達を弱めることを示し、上位脳の活性化が中脳水道周囲灰白質-吻側延髄腹内側部を介して慢性疼痛の増強に働いていると述べています。心の痛み(ストレス)でも同じような機序で慢性疼痛が増強すると考えています。
半場美智子先生の報告
1)機能的脳画像からみた慢性疼痛 –痛みの慢性化は予測可能か-
(ペインクリニック34:1100-1109, 2013)
半場先生は、2012年の総説でmesolimbic dopamine systemの機能低下が、慢性疼痛の痛覚過敏、意欲の低下を惹起する可能性について述べられたそうです。2013年の論文では①慢性疼痛の脳内では何が起きているのかの概略、②疼痛の慢性化は予測可能かについて述べられています。
①慢性疼痛の脳内では何が起きているのかの概略
慢性腰痛患者に熱刺激を加えて、脳の状態をMRIで調べると、健常者と変化はないそうです。しかし、その熱刺激を取り除いたときの脳の状態は、健常者と大きく異なっているそうです。健常者では、側坐核が熱刺激のonset時およびoffset時で、ともに応答が高まるそうですが、慢性疼痛患者では、onset時のみに反応し、offset時にはBOLD信号が健常者に比して減弱し、しかも自発痛を訴えるそうです。つまり、痛覚識別系に賦活がない場合にも自発痛を訴え、前頭前野と側坐核の機能的結合が高くなると述べています。PET検査では、慢性疼痛患者ではドーパミン代謝が低下し痛覚過敏に陥りやすいことも示されていることを紹介しています。
慢性腰痛患者では、外部刺激なしに自発痛が出現し、この時には前頭前野、前帯状回、側坐核が賦活され、視床や体性感覚野には賦活はみられていません。
②疼痛の慢性化は予測可能か
腰痛の慢性化の予測因子として、Apkarianらは、全脳スキャンの前頭前野-側坐核の機能的結合の大きさ、short form McGill pain questionnaire(sf-MPQ)のaffective高値をあげています。
つまり側坐核のBOLD信号、そして被験者の抑うつ気分などが予測因子として重要だということです。側坐核のactivityの低下は、痛みのkey componentということです。さらに側坐核はうつ病、睡眠障害、ストレスにも密接に関係していることを示しています。
2)痛みの新しい視点:mesolimbic dopamine system(ペインクリニック33:229-238, 2012)
Mesolimbic dopamine systemは快情動の中心と考えられてきましたが、痛みにも重要な役割を果たすことが明らかになってきました。慢性疼痛の患者ではこの経路に機能低下が示唆されると述べています。
腹側被蓋野(VTA)から側坐核や腹側淡蒼球などに向けてドーパミンが放出されます。ドーパミンが側坐核ニューロンを興奮させると、引き続きオピオイドが脳内のいろいろな神経核で分泌され快などを味わうそうです。このシステムが快情動ばかりでなく、痛みの制御にも関係していることが明らかにされてきたことを紹介しています。
最初に痛み誘発鎮痛の発現には、側坐核を中心としたドーパミンおよびオピオイドが働き、吻側延髄腹側部を介した下行性疼痛抑制系に関与することが示されています。
痛み刺激に対してのμ-オピオイド受容体の活性を紹介しています。高張食塩水で咬筋に注入し深部筋痛を起こし、その活性を調べた報告の紹介です。それでは、背側前帯状皮質、前帯状皮質膝周囲、内側前頭皮質、側坐核、腹側淡蒼球、扁桃体、島皮質、内側視床、中脳水道周囲灰白質に活性が起きることを示してあります。脳内オピオイド分泌がすすむと、被験者の疼痛は減少したそうです。
次に、末梢組織に痛み刺激が加わり、それからの一連の脳内応答を整理しています。ここは重要です。侵害情報は、脊髄後角より上行し、結合腕傍核、橋の脚橋被蓋野や背外側被蓋野を経て中脳の腹側被蓋野(VTA)に伝えられます。VTAから側坐核や腹側淡蒼球に向けてドーパミンが放出され(phasic activity)、側坐核ニューロンが興奮します。側坐核にはドーパミン受容体とオピオイド受容体が重なって豊富に存在し、側坐核のドーパミンによる興奮に引き続きオピオイド分泌が、側坐核、腹側淡蒼球、内側前頭皮質、扁桃体、島皮質、中脳水道周囲灰白質で生じ、下行性疼痛抑制系を介して、脊髄レベルで痛みが抑制されるということです。
一方、何週間も痛みが続いたり、ストレスが加わると、phasic activityは低下し、下行性疼痛抑制系が機能しなくなり、痛覚過敏状態が継続するそうです。不安、恐怖などが続くと、海馬内嗅皮質、前頭皮質、扁桃体からストレス性入力が加わり、VTAのphasic activityが低下すると述べています。
お二人の考えを私なりにまとめてみますと、頻回の疼痛やストレスにより、VTAから側坐核へのドーパミン放出(phasic activity)が低下するため、下行性疼痛抑制系の機能が低下し、慢性疼痛が惹起されると考えられるようです。
睡眠時の頭痛2014/01/01
一般に睡眠時の頭痛として、①群発頭痛、②片頭痛、③睡眠時頭痛、④睡眠時無呼吸による頭痛があげられます。群発頭痛や片頭痛については、各々のところで述べてきたので、簡単に触れます。睡眠時頭痛と睡眠時無呼吸による頭痛についても簡単にふれます。
各々の頭痛と睡眠との一般的な関係を示します。
| よくみられる頭痛のパターン・時間 | |
| 群発頭痛 | 午前零時から午前4時に多い。 |
| 片頭痛 | 夜中からおかしいなあと感じながら、起床時に頭痛。 |
| 睡眠時頭痛 | よく寝ていて、頭痛で目が覚める。 |
| 睡眠時無呼吸による頭痛 | 夜中にいびきをかき、呼吸は止まる。起床時に頭痛。 |
①群発頭痛
睡眠時の頭痛で、有名なのは群発頭痛です。夜、寝ついて1-2時間後に頭痛が起きて目が覚めますとか朝方4-5時頃に頭痛が出現しますなどという表現を患者様からよくお聞きします。群発頭痛は、年内周期と日内周期の二つの時間のサイクルがあると言われています。
以前紹介した2012年のアメリカからの報告を一部抜粋します。
【月別の群発頭痛】
群発頭痛が起きる月の頻度を示します。10月が最も多く26%、次いで9月4月3月11月となっています。秋と春ということでしょうか。しかし有意な差ではありません。
【時刻別の群発頭痛】
時刻別の頻度を示します。午前2時から3時というものが最も多く、午前0時から午前4時の間が多くなっています。
②片頭痛
睡眠時に片頭痛が起きて目を覚ますこともあるそうですが、むしろ多いのは、夜中からなんか変だなと感じながら、目が覚めてみると片頭痛発作がピークに達していたという状態です。起床時の片頭痛です。トリプタン製剤(マクサルトなど)は、痛みはじめに内服するという最良のタイミングを既に失っています。このような場合には、当院では、①トリプタン製剤の単独使用、②トリプタン製剤とNSAIDsなどの併用、③イミグラン皮下注(患者様自身で)、イミグラン点鼻などをおすすめしています。もちろん、軽度の場合は、NSAIDsやカロナールのみでもよいと思います。
また、睡眠不足や睡眠過多などが片頭痛の誘発因子ともなるので、一人一人の患者様自身が自分の片頭痛をみつめ、どのようなものが自身の片頭痛の誘発因子になっているのかを知り、それを予防することが大切です。
③睡眠時頭痛
睡眠時頭痛は、1988年にRaskinらによって報告された稀な頭痛です。頭痛は、睡眠時に起こり覚醒をきたします。
頭痛の程度は、中等度(moderate)から軽度(mild)のものが多いのですが、約20%で酷い(severe)頭痛をきたします。年齢は50歳以上で初発することが多く、持続時間は、通常は15分から180分です。多くの患者では、頭痛はほぼ毎日出現します。頭痛の性状は、締め付けられる頭痛、拍動性頭痛があります。
治療は、カフェイン、リチウム、インドメサシン、メラトニンが有効とされています。詳細は、→睡眠時頭痛へ
④睡眠時無呼吸性頭痛
ここでは閉塞性睡眠時無呼吸に目を向けます。1時間当たりの無呼吸低呼吸数apnea and hypopnea index (AHI)が5回以上を有意とし、日中の過眠などを伴うと睡眠時無呼吸症候群とします。
AHIが 5回-15回は軽度、15-30回は中等度、30回以上は重度とされています。
睡眠時無呼吸性頭痛は、ホメオスターシスによる頭痛に分類されています。
ICHD-3β版の診断基準を示します。
10.1.4 睡眠時無呼吸性頭痛の診断基準
- 頭痛は睡眠後の覚醒時に起こり、Cを満たす
- 睡眠時無呼吸(無呼吸-呼吸低下指数は5以上)が診断されている
- 原因となる証拠として、以下のうち少なくとも2項目が示されている
- 頭痛は、睡眠時無呼吸発作の発症と一致して発現している
- 以下のうち一方もしくは両方
- 頭痛は、睡眠時無呼吸発作の発症と並行して悪化した
- 頭痛は、睡眠時無呼吸発作が改善もしくは消失するのと並行して有意に改善したか消失している
- 頭痛は、以下の3項目 のうち、少なくとも1つを有する
- 1ヶ月に15日を超えて発現する
- 以下のすべて
(i) 両側 (ii)圧迫感 (iii)悪心、光過敏、音過敏を伴わない - 4時間以内に消失
- ほかに最適なICHD3の診断がない
起床時の頭痛を特徴とします。頭痛は、両側性、圧迫されるような頭痛とされています。
治療は、原因疾患の加療、減量、マウスピースの使用、CPAPなどです。
【症例のイメージ】
38歳女性。身長154㎝ 体重72㎏ BMI 30.3(肥満)
ほぼ毎日、朝起きてから頭痛があります。頭全体で圧迫されたような感じです。悪心はなく、光過敏や音過敏はありません。
AHI 60.1 最低SpO2 56。⇒睡眠時無呼吸症候群の重症例です。CPAPを開始。
CPAP治療を開始してからは、頭痛はありません。以前は、昼から夕にかけてカクンとくるような強烈な眠気がありましたが、治療をCPAP始めてからは、そういうことはなくなりました。